S-1/A: General form of registration statement for all companies including face-amount certificate companies
Published on June 1, 2023
As filed with the Securities and Exchange Commission on May 31, 2023
Registration No. 333-271699
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 2 to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Kiora Pharmaceuticals, Inc.
(Exact name of registrant as specified in its Charter)
| Delaware | 2834 | 98-0443284 | ||||||
|
(State or other jurisdiction of
incorporation or organization)
|
(Primary Standard Industrial
Classification Code Number)
|
(I.R.S. Employer
Identification No.)
|
||||||
332 Encinitas Boulevard, Suite 102, Encinitas, California 92024
(858) 224-9600
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive office)
Brian M. Strem, Ph.D.
President and Chief Executive Officer
Kiora Pharmaceuticals, Inc.
332 Encinitas Boulevard, Suite 102, Encinitas, California 92024
(858) 224-9600
(Name, address, including zip code, and telephone number, including area code, of agent for service)
With copies to:
| Robert A. Petitt, Esq. Burns & Levinson LLP 125 High Street Boston, MA 02110 (617) 345-3000 |
Michael Nertney, Esq.
Ellenoff Grossman & Schole LLP
1345 Avenue of the Americas
New York, New York 10105
(212) 370-1300
|
||||
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (check one)
| Large Accelerated filer | ☐ | Accelerated filer | ☐ | ||||||||
| Non-accelerated filer | ☒ |
Smaller reporting company | ☒ |
||||||||
| Emerging growth company | ☐ | ||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act .
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to Section 8(a), may determine.
SUBJECT TO COMPLETION, DATED MAY 31, 2023
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
PRELIMINARY PROSPECTUS

2,582,160 Shares of Common Stock
5,500 Shares of Series F Convertible Preferred Stock
(2,582,160 shares of Common Stock underlying the Series F Convertible Preferred Stock)
Class C Warrants to Purchase up to 2,582,160 Shares of Common Stock
(2,582,160 shares of Common Stock underlying the Class C Warrants)
and Class D Warrants to Purchase up to 2,582,160 Shares of Common Stock
(2,582,160 shares of Common Stock underlying the Class D Warrants)
We are offering 2,582,160 shares of common stock, including shares of common stock underlying shares of Series F Convertible Preferred Stock that we may issue as described below, together with Class C Warrants to purchase 2,582,160 shares of common stock (and the shares issuable from time to time upon exercise of the Class C Warrants) and Class D Warrants to purchase 2,582,160 shares of common stock (and the shares issuable from time to time upon exercise of the Class D Warrants) at an assumed combined purchase price of $2.13 per share of common stock, Class C Warrants and Class D Warrant pursuant to this prospectus. The shares and warrants will be separately issued but will be purchased together in this offering. Each warrant will have an exercise price of $ per share and will be exercisable upon issuance. The Class C Warrants and the Class D Warrants will include a one-time reset of the exercise price to a price equal to the lesser of (i) the then exercise price and (ii) 90% of the five-day volume weighted average prices for the five (5) trading days immediately preceding the date that is sixty calendar days after issuance of the Class C Warrants and Class D Warrants. The Class C Warrants will expire five years from the date of issuance and the Class D Warrants will expire one year from the date of issuance.
We are also offering to those purchasers, whose purchase of shares of common stock in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock following the consummation of this offering, the opportunity to purchase, if they so choose, in lieu of the shares of our common stock that would result in ownership in excess of 4.99% (or, at the election of the purchaser, 9.99%), shares of Series F Convertible Preferred Stock (“Series F Preferred Stock”), convertible at any time at the holder’s option into a number of shares of common stock equal to $1,000 divided by the combined public offering price (the “Conversion Price”), at a public offering price of $1,000 per share of Series F Preferred Stock. Each share of Series F Preferred Stock is being sold together with the same warrants described above being sold with each share of common stock. For each share of common stock underlying a share of Series F Preferred Stock that we sell, the number of shares of common stock that we are selling will be decreased on a one-for-one basis.
The price of our common stock on The Nasdaq Capital Market during recent periods will only be one of many factors in determining the final combined public offering price. Other factors to be considered in determining the final combined public offering price include our history, our prospects, the industry in which we operate, our past and present operating results, the general condition of the securities markets at the time of this offering and discussions between the underwriters and prospective investors. The recent market price used throughout this prospectus may not be indicative of the final combined public offering price. All share and warrant numbers included in this prospectus are based upon an assumed combined public offering price per share of common stock, Class C Warrant and Class D Warrant of $2.13, the closing price of our common stock on The Nasdaq Capital Market on May 30, 2023.
Our common stock is listed on The Nasdaq Capital Market under the symbol “KPRX.” On May 30, 2023, the last reported sale price of our common stock on The Nasdaq Capital Market was $2.13 per share. The Class C Warrants, Class D Warrants and any shares of Series F Preferred Stock that we issue are not and will not be listed for trading on The Nasdaq Capital Market.
You should read this prospectus, together with additional information described under the headings “Incorporation of Certain Information by Reference” and “Where You Can Find More Information,” carefully before you invest in our common stock.
Investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 18 of this prospectus, and under similar headings in the other documents that are incorporated by reference into this prospectus, for a discussion of information that should be considered in connection with an investment in our securities.
Neither the U.S. Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Per Share and Warrants(1)
|
Total | |||||||||||||
| Public offering price | $ | $ | ||||||||||||
Underwriter discounts and commissions (2)
|
$ | $ | ||||||||||||
| Proceeds, before expenses, to us | $ | $ | ||||||||||||
__________________
(1)The public offering price and underwriting discount corresponds to (i) a public offering price per share of common stock of $ ($ net of the underwriting discount) and (ii) a public offering price per warrant of $ ($ net of the underwriting discount).
(2)We have agreed to pay certain expenses of the underwriters in this offering. We refer you to “Underwriting” on page 64 for additional information regarding underwriting compensation.
The offering is being underwritten on a firm commitment basis. We have granted a 45-day option to the underwriters to purchase up to an additional 387,324 shares of common stock, Class C Warrants to purchase up to an additional 387,324 shares of common stock and/or Class D Warrants to purchase an additional 387,324 shares of common stock at the public offering price, less the underwriting discounts payable by us, to cover over-allotments, if any.
The underwriters expect to deliver the securities to investors on or about , 2023.
Sole Book-Running Manager
Ladenburg Thalmann
The date of this prospectus is , 2023
TABLE OF CONTENTS
DESCRIPTION OF THE SECURITIES WE ARE OFFERING
|
|||||
ABOUT THIS PROSPECTUS
We have not, and the underwriters have not, authorized anyone to provide you with information that is different from that contained in this prospectus or in any free writing prospectus we may authorize to be delivered or made available to you. When you make a decision about whether to invest in our securities, you should not rely upon any information other than the information contained in or incorporated by reference in this prospectus or in any free writing prospectus that we may authorize to be delivered or made available to you. Neither the delivery of this prospectus nor the sale of our securities means that the information contained in this prospectus or any free writing prospectus is correct after the date of this prospectus or such free writing prospectus. This prospectus is not an offer to sell or the solicitation of an offer to buy our securities in any circumstances under which the offer or solicitation is unlawful.
For investors outside the United States: We have not, and the underwriters have not, taken any action that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the securities covered hereby and the distribution of this prospectus outside the United States.
Unless otherwise indicated, information contained in this prospectus concerning our industry and the markets in which we operate, including our general expectations and market position, market opportunity and market share, is based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable. Our management estimates have not been verified by any independent source, and we have not independently verified any third-party information. In addition, assumptions and estimates of our and our industry’s future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors.” These and other factors could cause our future performance to differ materially from our assumptions and estimates. See “Special Note Regarding Forward-Looking Statements.”
We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to the registration statement of which this prospectus is a part were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and covenants should not be relied on as accurately representing the current state of our affairs.
We have proprietary rights to trademarks used in this prospectus, including Kiora®. Solely for our convenience, trademarks and trade names referred to in this prospectus may appear without the “®” or “™” symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent possible under applicable law, our rights or the rights to these trademarks and trade names. We do not intend our use or display of other companies’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, any other companies. Each trademark, trade name, or service mark of any other company appearing in this prospectus is the property of its respective holder.
i
PROSPECTUS SUMMARY
The following summary highlights information contained elsewhere in this prospectus. It may not contain all of the information that is important to you. You should read the entire prospectus carefully, especially the discussion regarding the risks of investing in our securities under the heading “Risk Factors,” before investing in our securities. All references to “Company” “we,” “our” or “us” refer solely to Kiora Pharmaceuticals, Inc. and its subsidiaries and not to the persons who manage us or sit on our Board of Directors.
Overview
We are a clinical-stage specialty pharmaceutical company developing and commercializing therapies for the treatment of ophthalmic diseases. We were formed as a Delaware corporation on December 26, 2004, under the name of EyeGate Pharmaceuticals, Inc., and changed our name to Kiora Pharmaceuticals, Inc. effective November 8, 2021. We were originally incorporated in 1998 under the name of Optis France S.A. in Paris, France. Our lead product is KIO-301, with an initial focus on patients with later stages of vision loss due to retinitis pigmentosa (RP, any and all sub-forms). KIO-301 is a potential vision-restoring small molecule that acts as a “photoswitch” specifically designed to restore vision in patients with inherited and age-related degenerative retinal diseases, including RP. The molecule is designed to restore the eyes’ ability to perceive and interpret light in visually impaired patients through selectively entering viable downstream retinal ganglion cells (no longer receiving electrical input due to degenerated rods and cones) and turning them into light sensing cells, capable of signaling the brain as to the presence or absence of light. We initiated a Phase 1b clinical trial in the third quarter of 2022 and dosed the first patient in November 2022. On March 17, 2022, we were granted orphan drug designation (ODD) by the United States Food and Drug Administration (FDA) for the active pharmaceutical ingredient (API) in KIO-301. KIO-301 (formerly known as B-203) was acquired through the Bayon Therapeutics, Inc. (Bayon) transaction which closed October 21, 2021.
KIO-101 focuses on treating the ocular manifestation of patients with autoimmune diseases, including rheumatoid arthritis and, as such, is termed the Ocular Presentation of Rheumatoid Arthritis and Other Autoimmune Diseases (OPRA+). KIO-101 is a next-generation, non-steroidal, immuno-modulatory, small-molecule inhibitor of dihydroorotate dehydrogenase (DHODH). We believe KIO-101 to be best-in-class with picomolar potency and a validated immune modulating mechanism designed to overcome the off-target side effects and safety issues associated with commercially available DHODH inhibitors. In the fourth quarter of 2021, we reported top-line safety and tolerability data from a Phase 1b proof-of-concept (POC) study evaluating KIO-101 in patients with ocular surface inflammation. As a further sign of safety, there were zero clinically significant laboratory (including liver enzymes) findings observed in both healthy patients and those with ocular surface inflammation. We initiated a Phase 2 clinical trial in the first quarter of 2023. KIO-101 (formerly known as PP-001) was acquired through the acquisition of Panoptes Pharma GmbH (Panoptes) in the fourth quarter of 2020.
We are developing KIO-201 for patients with persistent corneal epithelial defects (PCED). PCED is an orphan disease and as such, we are currently seeking orphan drug designation (ODD). KIO-201 is also being considered for patients recovering from surgical wounds, such as those undergoing the laser vision correction procedure, photorefractive keratectomy (PRK). KIO-201 is a modified form of the natural polymer hyaluronic acid, designed to protect the ocular surface to permit re-epithelialization of the cornea and improve and maintain ocular surface integrity. KIO-201 has unique properties that help hydrate and protect the ocular surface. We completed a Phase 2 clinical trial in patients with PCEDs and released top-line data in Q1 2023. We expect to release full data in Q2 2023. We are in planning stages of a Phase 3b trial for patients recovering from PRK and plan to initiate the study before the end of 2023.
1
Market Opportunity
Retinitis Pigmentosa Market Overview
More than 5.5 million patients globally are estimated to have an inherited retinal disease leading to significant or permanent vision loss. RP is the largest family of these inherited diseases. RP affects about 1 in 3,500 people worldwide. Thus, with a population of about 330 million in the U.S. as of February 2021, about 96,250 people in the U.S. would be expected to have RP. With a worldwide population presently estimated at over 7.7 billion, it can be estimated that approximately 2.3 million people around the world have RP.
RP is a group of hereditary progressive disorders that may be inherited as autosomal recessive, autosomal dominant or X-linked recessive traits. Maternally inherited variants of RP transmitted via the mitochondrial DNA can also exist. About half of all RP cases are isolated (that is, they have no family history of the condition). RP may appear alone or in conjunction with one of several other rare disorders. Patients with RP have a progressive loss of photoreceptors (rods and cones) and therefore patients with late-stage RP have a substantial loss of peripheral and central visual function.
While no approved therapies are available for the treatment of RP, current therapeutics in development primarily rely on genetic approaches to introduce light sensing channels into viable downstream cells, a field termed optogenetics. KIO-301 is a small molecule photoswitch, that confers light sensitivity to downstream cells, specifically the retinal ganglion cells (RGCs), potentially triggering the same phototransduction signaling as if the photoreceptors were present and viable.
Our Solution: KIO-301
KIO-301 is a novel small molecule with the potential to confer light sensitivity to patients with degenerated retinas due to either inherited or age-related diseases, which has received orphan drug designation from the FDA. Many retinal diseases result in the death of the retinal photoreceptors, the light sensing cells in the retina. However, downstream retinal neurons, such as the bipolar and RGCs remain viable for long periods after photoreceptor death. KIO-301 selectively enters these cells and non-covalently resides on the intracellular domains of potassium and hyperpolarization-activated, cyclic nucleotide-gated (HCN) voltage gated ion channels. As KIO-301 has an azobenzene core, visible light causes a rapid and reversible change in the isomeric state of the molecule, transforming from a linear molecule to an orthogonal molecule. When this happens, the voltage gated ion channels and current efflux are blocked, causing cellular depolarization and signaling to the brain as to the presence of light. When light is no longer touching the molecule, it reverts back to its linear state, allowing ion efflux from the cells and thus promoting repolarization and a turning “off” of the brain signaling.
This novel mechanism of action enables potential application to multiple diseases. RP is a group of inherited eye diseases that cause photoreceptor cell death. In the U.S., RP is considered an orphan disease with a prevalence of fewer than 200,000. This prevalence enables consideration for KIO-301 to qualify for orphan drug designation in the treatment of RP, conferring increased regulatory collaboration with the FDA, and market exclusivity if clinical trials demonstrate safety and efficacy. On March 17, 2022, we were granted orphan drug designation by the FDA for the active ingredient in KIO-301. Currently, no therapeutics are approved to treat patients with RP.
A possible market expansion from RP would be to evaluate KIO-301 in patients with geographic atrophy (GA), the late stage of age-related dry macular degeneration. There are about 1,000,000 patients in the U.S. with GA and to date, only one therapeutic has been approved to treat this disease (Syfovre) which is intended to slow the disease progression but not return vision that was lost due to disease.
Ocular Presentation of Rheumatoid Arthritis Market Overview
Patients with systemic autoimmune diseases including Rheumatoid Arthritis (RA), are known to suffer from ocular presentation of their underlying autoimmune conditions. Secondary to inflammation and associated pathologies in the joint synovium, the eye carries significant morbidity and impact on eye health and quality of life. These ocular presentations can include signs and symptoms similar to keratoconjunctivitis sicca (KCS), episcleritis, scleritis, peripheral ulcerative keratitis (PUK), anterior uveitis, as well as retinal vasculitis. In patients with OPRA+,
2
the surface of the eye often has significant irritation accompanied by symptoms of soreness, grittiness, light sensitivity, and dryness. Patients with RA suffer from ocular signs and symptoms at a rate reported to be 2-3X that of the general population. Furthermore, in those OPRA+ patients, up to 50% report moderate to severe signs and symptoms. Today, there are approximately 1.8 million RA patients in the U.S. Approximately one-third of these patients present with OPRA+ (more than 0.5 million in the U.S.), with more than 90% seeking prescription medication to address these ophthalmic manifestations. Unfortunately, today's ocular surface anti-inflammatory medicines are usually not sufficient to treat OPRA+ as they are broad and not targeted to the underlying pathophysiology.
As noted above, KIO-101 is a member of a family of DHODH inhibitors, known to be disease modifying agents in autoimmune diseases. RA, as well as OPRA+, are T-cell mediated auto-inflammatory diseases and whilst rheumatologists are helping the systemic manifestations of this disease with approved targeted t-cell modulators, including DHODH inhibitors, ophthalmologists do not have the same toolbox of treatments designed specifically to help patients with ocular presentation.
Our Solution: KIO-101
KIO-101 is a third-generation small molecule DHODH inhibitor. DHODH is extensively exploited as potential drug targets for immunological disorders, oncology, and infectious diseases. DHODH is a key enzyme in the de novo pyrimidine synthesis pathway. This enzyme is located in the mitochondria and catalyzes the conversion of dihydroorotate (DHO) to orotate as the fourth step in the de novo synthesis of pyrimidines that are ultimately used in the production of nucleotides.
Nucleotides are required for cell growth and replication. Nucleotides are the activated precursors of nucleic acids and are necessary for the replication of the genome and the transcription of the genetic information into RNA. Nucleotides also serve as an energy source for a more select group of biological processes (ATP and GTP). They also play a role in the formation of glycogen, signal-transduction pathways, and as components of co-enzymes (NAD and FAD). An ample supply of nucleotides in the cell is essential for all cellular processes.
There are two pathways for the biosynthesis of nucleotides: salvage and de novo. The main difference is where the nucleotide bases come from. In the salvage pathway, the bases are recovered (salvaged) from RNA and DNA degradation. In the de novo pathway, the bases are assembled from simple precursor molecules (made from scratch).
One critical requirement of fast-growing or proliferating cells, such as the expansion of activated B- and T-cells, cancer cells, and pathogen infected host cells, is the requirement of an abundance of nucleotide bases. These metabolic activities will predominately utilize the de novo pathway for nucleotide biosynthesis. A key advantage of DHODH inhibition is the selectivity towards metabolically activated cells (with a high need for RNA and DNA production), which should mitigate any negative impact on normal cells. Depletion of cellular pyrimidine pools through the selective inhibition of DHODH has been shown to be a successful approach for therapeutic development.
Currently, two first generation DHODH inhibitors have been approved in the U.S. and abroad and are marketed by Sanofi as leflunomide (Arava®) and the active metabolite teriflunomide (Aubagio®). These oral tablets are approved for the treatment of rheumatoid and psoriatic arthritis and multiple sclerosis (MS), respectively. These diseases are autoimmune disorders. One potential explanation for the therapeutic effects of Arava® in arthritis is the reduction in the numbers or reactivity of activated T-cells, which are involved in the pathogenesis of arthritis. The generally accepted view of human MS pathogenesis implicates peripheral activation of myelin-specific autoreactive T-cells that lead to inflammatory disease in the central nervous system (CNS). By blocking the de novo pyrimidine synthesis pathway via DHODH inhibition, it is suggested that Aubagio® reduces T-cell proliferation in the periphery. Arava® and Aubagio® are formulated as oral drugs and it is established that leflunomide will be metabolized in the liver to the active metabolite teriflunomide. Hepatotoxicity was reported as a major side effect after oral administration, possibly as a result of the extent of liver metabolism. Moreover, it was shown that apart from DHODH, a series of protein kinases are inhibited by Arava® and Aubagio®.
3
Corneal Wounds Market Overview
Normal corneal epithelial wound healing relies on rapid migration and proliferation of epithelial cells from the wound edge and the limbus, followed by extracellular matrix deposition and remodeling. Persistent corneal epithelial defects (PCED) are corneal wounds, caused by injury or disease, that do not heal within the normal time frame (usually 7-14 days) but persist for weeks or even months. Several underlying disease states may result in PCED, including previous herpes simplex or herpes zoster infection, neurotrophic keratitis, diabetes, and severe dry eye states. Nonhealing corneal epithelial defects may also occur after ocular surgery or other physical injuries and/or trauma to the cornea. PCEDs require intervention as they can lead to infections, stromal ulceration, corneal scarring, and opacification and result in vision loss.
There is an unmet medical need for a simple treatment that could aid in the healing of PCEDs. Current methods of addressing PCEDs include debridement and patching, applying a bandage contact lens, human amniotic membrane, autologous serum, suturing the lids via a tarsorrhaphy, or in severe cases applying a conjunctival graft over the cornea. These methods are invasive, costly, and/or merely cover the wound; none have proven universally effective for healing PCEDs and often result in a recurrence of the defect. KIO-201, a crosslinked, modified hyaluronic acid, is applied topically as a gel eye drop formulation and works to stabilize the tear film and protect the ocular surface, providing a local tissue environment conducive to re-epithelialization and corneal wound healing.
Further, there are multiple surgical procedures involving the ocular surface that have long recovery, whereby acceleration of that period would benefit the patients. PRK surgery is an efficacious alternative to patients seeking surgical correction of refractive errors who are not suitable candidates for Laser-Assisted In Situ Keratomileusis (LASIK) due to inadequate corneal thickness, larger pupil size, history of KCS, or anterior basement membrane disease. PRK surgery involves controlled mechanical removal of corneal epithelium with subsequent excimer laser photoablation of the underlying Bowman’s layer and anterior stroma, including the subepithelial nerve plexus.
The military prefers PRK as a refractive procedure due to the stability of the PRK incision and the absence of risk for flap dislocation during military active duty. Although this procedure yields desirable visual acuity results, common complications of the procedure include post-operative pain secondary to the epithelial defects, risk of corneal infection prior to re-epithelization of the large epithelial defect, corneal haze formation, decreased contrast sensitivity, and slower visual recovery. The number of laser vision correction procedures is on the rise, estimated in 2021 at about 2 million in the U.S.. Whilst PRK comprises a fraction of these procedures, there are about 160,000 surgeries performed annually in the U.S. These surgeries are heavily consolidated to a few corporate umbrellas, such as TLC Laser Eye Centers, enabling a targeted commercial campaign once a therapeutic is approved.
Keratoconus is an orphan disease of the ocular surface, affecting approximately 170,000 patients in the U.S. alone according to NORD. Keratoconus progression involves the structure of the cornea which bulges outward, directly affecting vision. Whilst the etiology of the disease is unknown, there are multiple approaches to helping these patients, involving the use of vision correction prothesis such as contact lenses and glasses, to surgical approaches involving collagen cross-linking the corneal surface to provide more rigidity and slow progression. One of these corneal cross-linking approaches, termed epi-off, involves the removal of about 8 mm of the epithelium on the cornea and a riboflavin solution is applied to the exposed corneal stroma. This procedure is not free of side effects, which often include corneal infections, subepithelial haze, sterile infiltrates, reactivation of herpetic keratitis, and endothelial damage. Thus, accelerating the re-epithelialization would carry significant value.
Our Solution: KIO-201
KIO-201 is a synthetic modified hyaluronic acid (HA) capable of coating the ocular surface and designed to resist degradation under conditions present in the eye. This prolongs residence time of the bandage on the ocular surface, thereby addressing one of the limitations of current non-cross-linked HA formulations. Additionally, cross-linking allows the product’s viscosity to be modified to meet optimum ocular needs. The improved viscoelasticity and non-covalent muco-adhesive interfacial forces improve residence time in the tear film, thus providing a coating that aids re-epithelization of the ocular surface via physical protection. If KIO-201 is approved by the FDA, we expect that it will be the only wound healing prescription eye drop available in the U.S. based on HA.
4
KIO-201 exhibits significant shear thinning properties. This feature allows the modified HA to act as a more concentrated, viscous barrier at low shear rates in a resting tear film, but also as a lower resistance fluid (therefore thinned) during high shear events such as blinking. This property enables better residence time and a more favorable ocular surface coating with less optical blur. We have demonstrated in animal studies that KIO-201 remains on the ocular surface for up to two hours, and further demonstrated in a human clinical study that KIO-201 does not cause blurriness while on the ocular surface. This enhances ocular surface protection and patient comfort, while maintaining good visual function.
KIO-201 has been shown to provide a mechanical barrier that aids in the management of corneal epithelial defects and re-epithelization in both preclinical studies and in clinical ophthalmic use. Patients with PCEDs or those recovering from PRK surgery provide subject populations in need of safe and efficacious treatments to accelerate recovery. KIO-201 has already demonstrated statistical significance in a Phase 2a PCED clinical trial and in a pivotal clinical study for its ability to accelerate wound healing against the current standard-of-care, a bandage contact lens.
A possible market expansion would be to evaluate KIO-201 in patients with keratoconus, an ocular disease that affects the structure of the cornea and can cause blindness. Currently patients undergo a mechanical reshaping of the cornea, however this approach causes significant damage to the epithelial layer. As KIO-201 has demonstrated the ability to accelerate wound healing to the ocular surface, the underlying mechanism of action would be a congruent fit.
Our Strategy
Our goal is to develop products for treating disorders of the eye. The key elements of this strategy are to:
•Develop Core Assets
–Continue clinical development of KIO-301 in a Phase 1b clinical study in patients with mid to late stage retinitis pigmentosa.
–Continue clinical development of KIO-101 in a Phase 2 clinical study for the treatment of the ocular manifestations of autoimmune diseases (e.g., rheumatoid arthritis). In the first quarter of 2023, we received approval to initiate a Phase 2 study, slated to start enrollment in the second quarter of 2023.
–Continue clinical development of KIO-201 in patients with PCEDs and consider advancing our late stage program for patients undergoing surgical vision correction including PRK and certain types of keratoconus surgical procedures. These programs will benefit from discussions with the FDA regarding clinical trial designs and approvable endpoints.
•Expand Portfolio through Collaborations
–Pursue strategic collaborations to further the Company’s existing assets with respect to new indication potential and more detailed mechanism of action, which can result in new intellectual property.
5
Our Development Pipeline
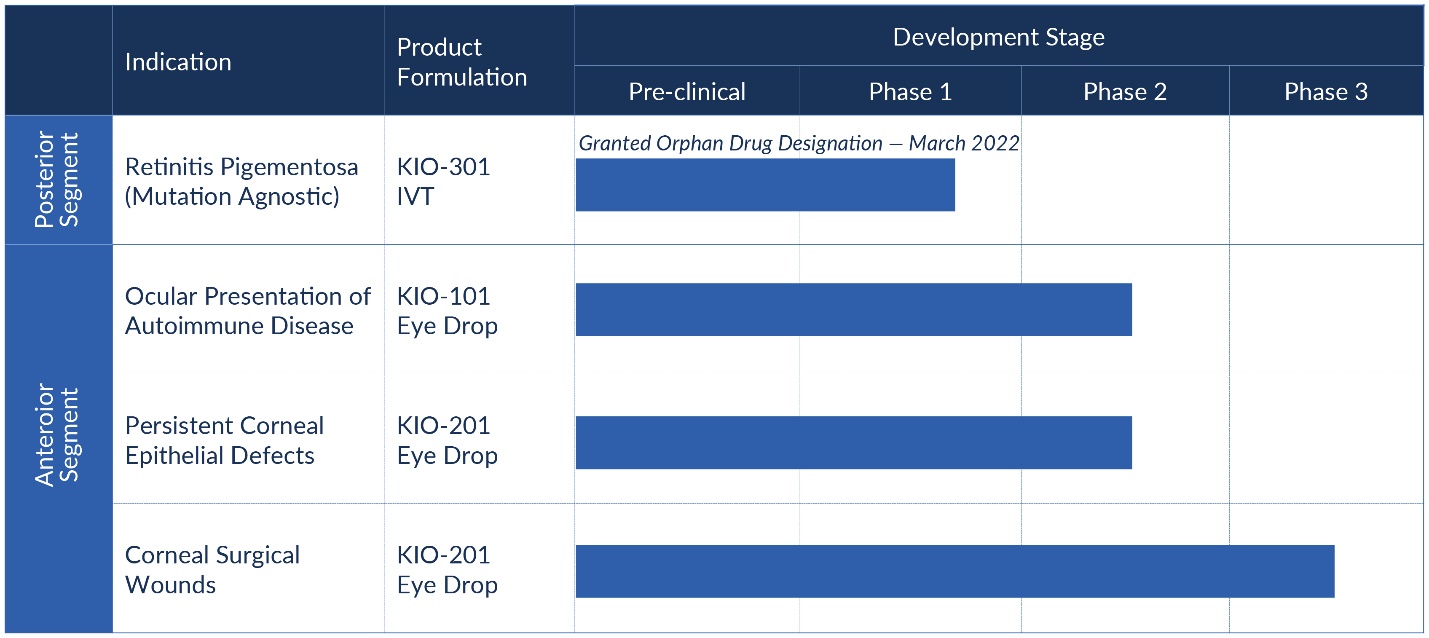
Clinical Development
KIO-101: Ocular Presentation of Rheumatoid Arthritis and Other Autoimmune Diseases (OPRA+)
Mechanism of Action
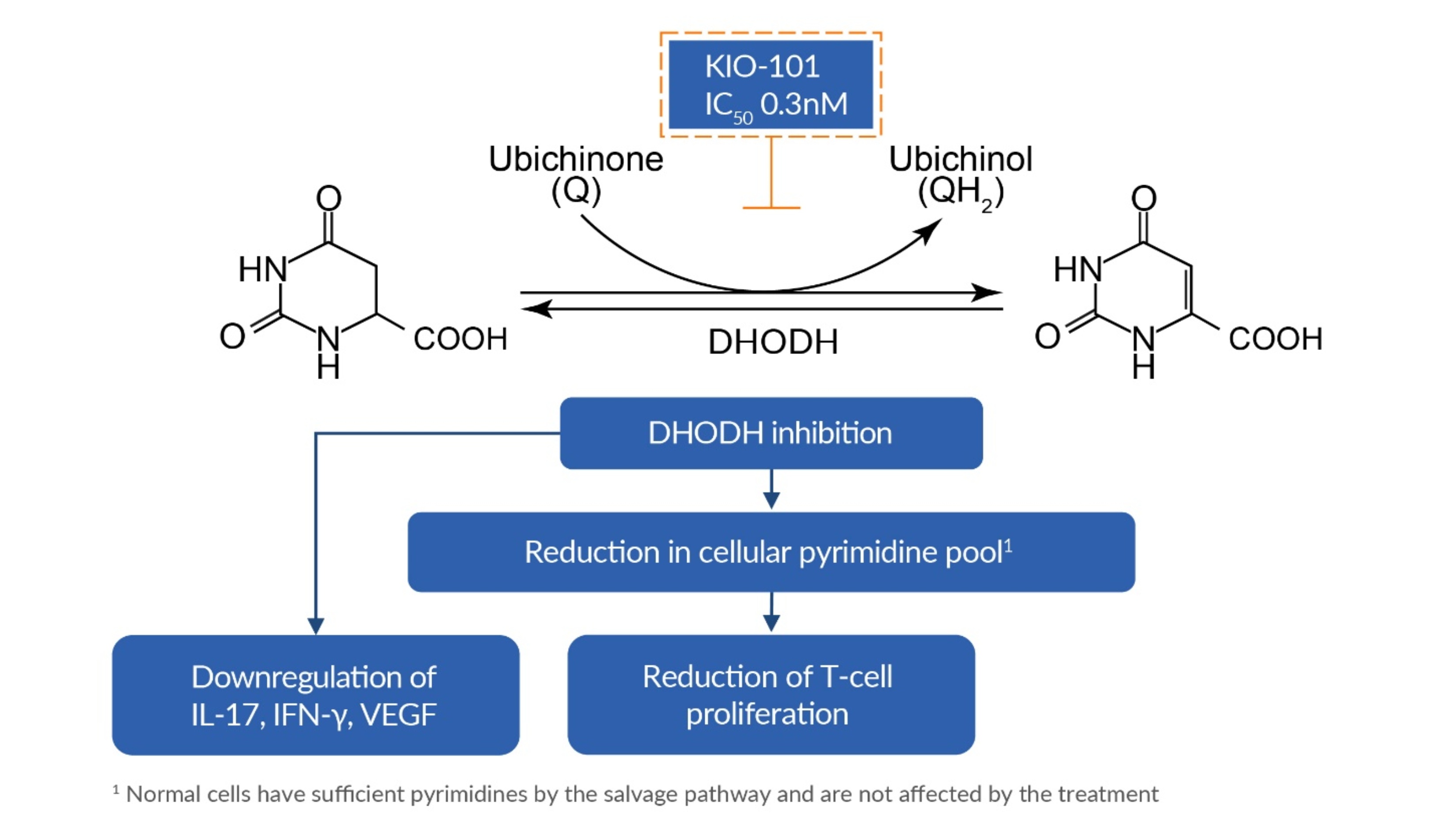
KIO-101 is a promising novel third generation DHODH inhibitor, with a half-maximal inhibitory concentration IC50-value of 0.3 nM. Based on internal work completed, we believe this means that 1,000-fold more potent than teriflunomide (IC50 DHODH 415 nM). Furthermore, KIO-101 suppresses the expression of key pro-inflammatory cytokines such as IL-17, IFN-g, VEGF and others, potentially as a consequence of inhibiting DHODH. IL-17 and IFN-g are the hallmark cytokines expressed by Th1 and Th17 T-cells, respectively, and play a crucial role in initiating the inflammatory processes in several ocular diseases, including dry eye disease (including the association with autoimmune conditions such as rheumatoid arthritis) and non-noninfectious uveitis. KIO-101 is structurally and mechanistically different from Arava®, a drug currently approved by the FDA for the treatment of rheumatoid arthritis. The IC50 of KIO-101 on selected tyrosine kinases, such as PI3K, AKT and JAK, is more than 10,000-fold above the IC50 of KIO-101 for DHODH. In general, side effects are not expected and have not been observed to date in animal and human studies after KIO-101 administration
6
The postulated mode of action of KIO-101 is depicted below.
Phase 1b Study:
The results of a Phase 1b study of KIO-101 eye drops in adults with and without ocular surface inflammation were reported in Q4 2021.
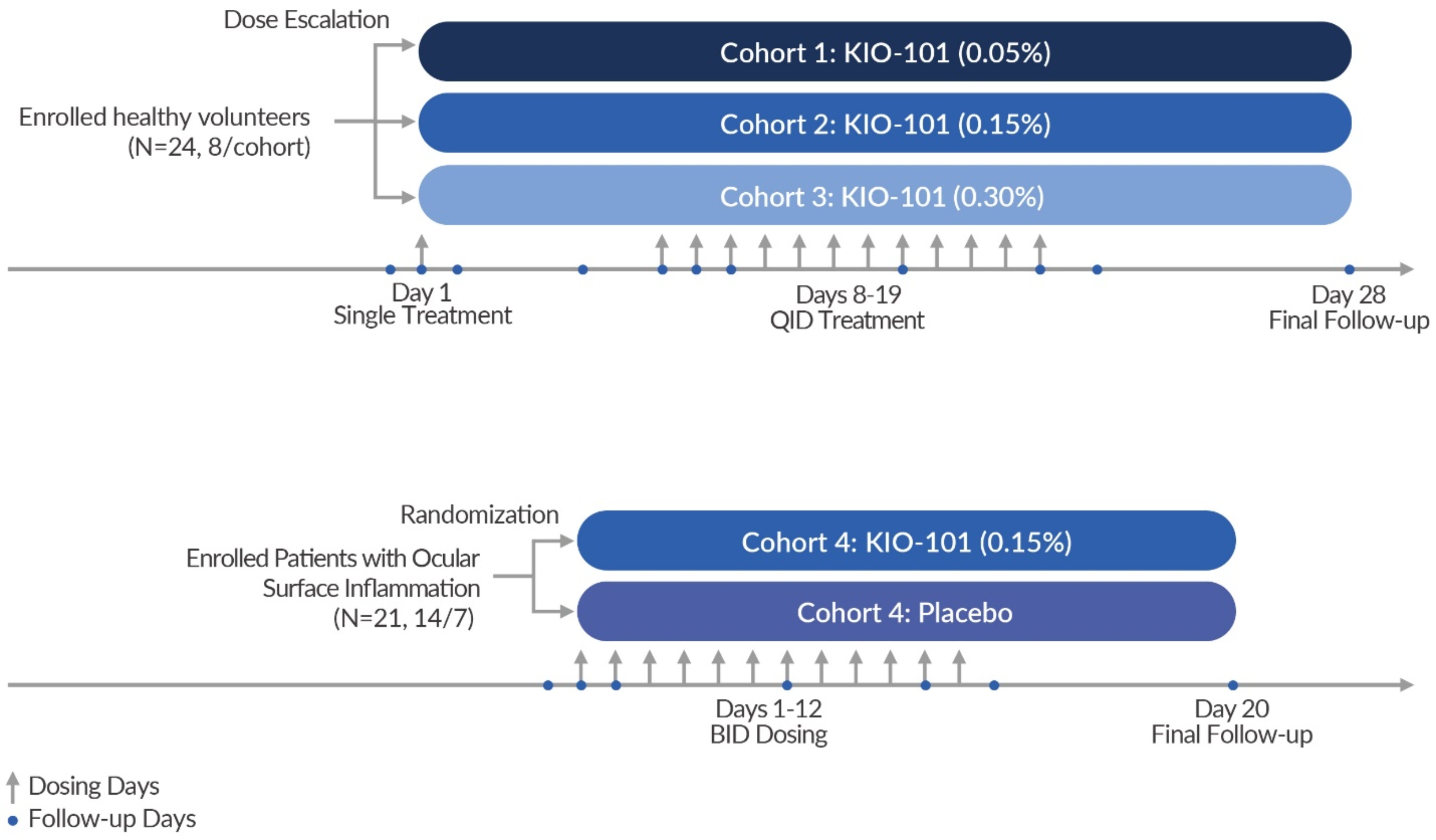
Design
The first part of this single center, randomized, double-masked study was to explore safety and tolerability of KIO-101 in a healthy population; and the second part was to investigate a potential efficacy signal in patients with ocular surface inflammation and hyperemia. Part 1 (cohorts 1 through 3) consisted of healthy volunteers receiving dose escalating concentrations of KIO-101 as noted on the figure below. Specifically, healthy volunteers were repeatedly treated with ascending doses of KIO-101 (0.05%, 0.15%, 0.30%) and placebo eyedrops. Subjects receiving 0.05% and 0.15% eyedrops showed excellent tolerability. Both doses can be used for future studies in patients having an infection or inflammation on the ocular surface. No Severe Adverse Events (SAEs) or severe ocular Adverse Events (AEs) were reported in any patients. In the 0.3% group, two patients withdrew for epistaxis and further dosing in the entire 0.3% group was stopped. No lab abnormalities in these two or any patients were observed and further toxicology studies are ongoing, including the 0.3% dose.
In the second part (cohort 4) of this study, 21 patients diagnosed with ocular surface inflammation, a key driver of ocular surface disease including dry eye disease, were evaluated. These patients were treated BID for 12 days with 0.15% of KIO-101 (n=14) or vehicle (n=7). The key inclusion criteria were conjunctival hyperemia score >2 (on the Efron scale of 0-5) and an Ocular Surface Disease Index© (OSDI) score of > 22. Primary endpoints included safety and tolerability. Secondary and exploratory endpoints included pharmacokinetics of KIO-101 as well as
7
change from baseline in OSDI, conjunctival hyperemia, tear break up time (TBUT), corneal staining (fluorescein), conjunctival staining (lissamine green), ocular discomfort, lid edema, and lid erythema.
Study Results
The results demonstrated favorable safety and tolerability of KIO-101, as well as statistically significant improvements in conjunctival hyperemia, a key inclusion criterion for the 21 patients enrolled with ocular surface inflammation and a recognized clinical sign in patients with ocular surface inflammation. At Day 13, 100% (Figure 1 below) of patients treated with KIO-101 (14/14) saw a reduction >1 from baseline, measured on the Efron scale (0-5), versus only 42.8% with vehicle control (3/7) (p < 0.006). The mean reduction in conjunctival hyperemia score from baseline to Day 13 demonstrated statistically significant difference in active treatment vs. vehicle control groups (-1.055 vs. -0.604; p = 0.0316). This apparent drug effect on conjunctival hyperemia was lost when patients were assessed at the Day 20 post-treatment follow-up, which occurred 8 days after the last dose was administered, further supporting a potential positive drug effect. There was a numerical trend favoring KIO-101 in OSDI, but no statistically significant differences were observed in TBUT, corneal staining, conjunctival staining, or other
8
exploratory endpoints. A larger sample size and dosing period longer than two weeks will likely be necessary to effectively evaluate a statistical drug effect on these additional efficacy endpoints.
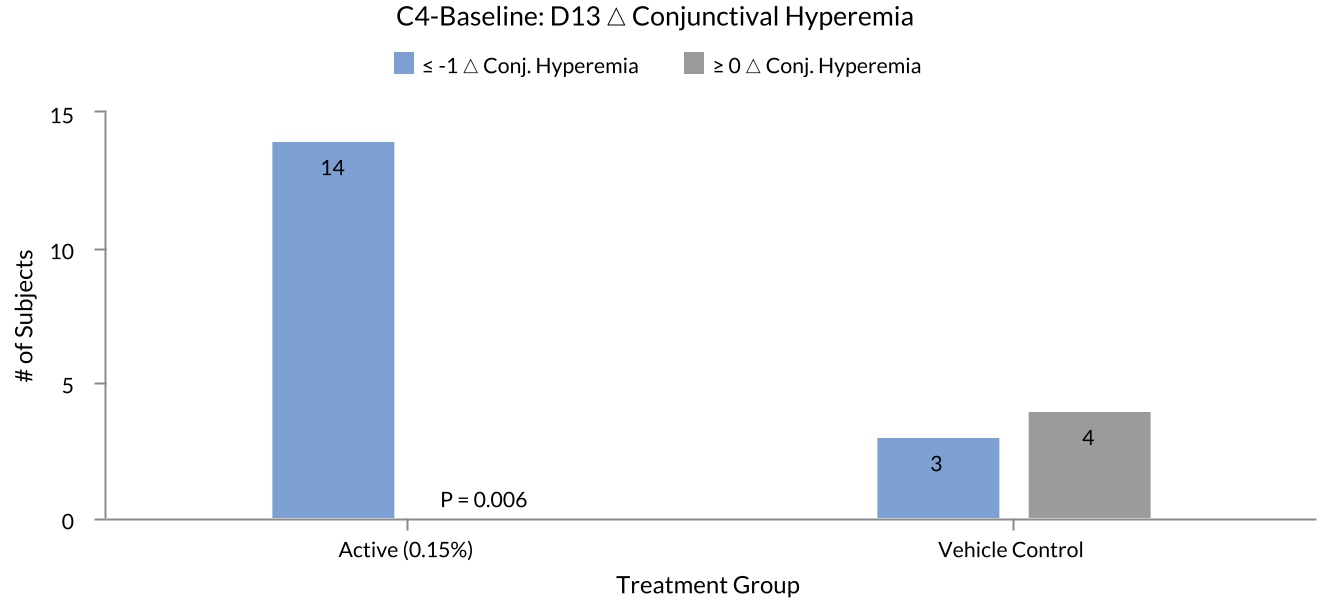
Figure 1: Percent of patients with reduction of >1
No SAEs or severe ocular AEs were reported. In the 0.3% group, 2 patients withdrew for epistaxis (nose bleeds) and dosing was stopped, with no lab abnormalities in these 2 or any patients observed. In cohort 4, no difference was observed in the frequency of ocular AEs in active vs. control.
Clinical Development Plan
In February 2023 we received investigational new drug application approval for a Phase 2 study of KIO-101 for the treatment of OPRA+ and initiated enrollment in Q2 2023. The study will enroll approximately 120 patients in a multi-center, controlled, randomized, double-masked trial assessing the safety and efficacy of KIO-101 eye drops in patients living with autoimmune disease who have signs and symptoms of ocular surface disease.
KIO-201: Persistent Corneal Epithelial Defects
Phase 2 Study:
In Q1 2022, we initiated a clinical trial of KIO-201 evaluating the potential to help patients with PCEDs. We completed this study in Q4 2022 and are presenting the results in Q2 2023.
Design
This single site clinical trial was designed to enroll up to 10 patients (20 eyes) with PCEDs, as defined by a duration of at least 14 days while on conventional therapies. The primary endpoint of the study is safety and tolerability with key secondary endpoints including assessing the number of patients with healing (defined by lesions being <0.5mm2).
Clinical Development Plan
PCED qualifies as a rare disease, and therefore we applied for orphan drug designation as part of our clinical development plan for KIO-201 in Q4 2022. We expect to pursue ODD in 2023 and with the results from the aforementioned clinical trial, we plan to meet with the FDA to discuss next steps.
9
KIO-201: PRK Surgical Recovery Pivotal Study
Pivotal Study:
In Q4 2019, we reported positive topline results from our corneal wound repair pivotal clinical trial of KIO-201 for the corneal re-epithelialization in patients having undergone PRK surgery.
Design
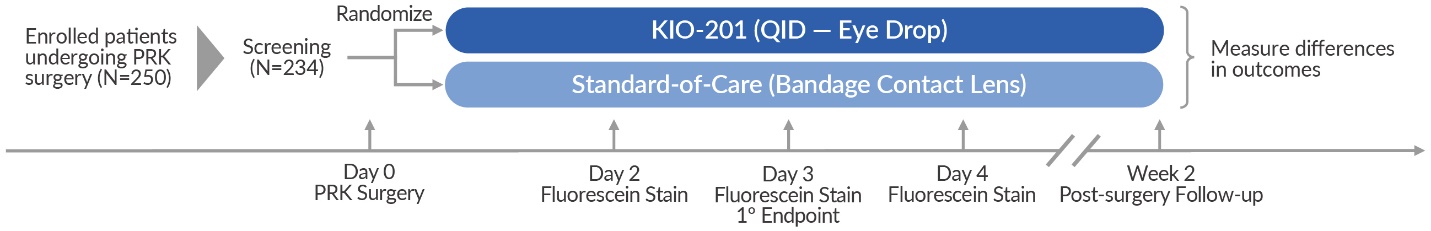
The prospective, controlled study randomized 234 patients undergoing bilateral PRK surgery and was designed to assess safety and efficacy by comparing KIO-201 to the current standard-of-care, a bandage contact lens (BCL). The primary endpoint was the proportion of study eyes achieving complete wound closure on Day 3 (and remaining closed). This assessment was evaluated by an independent masked reading center, using digital slit-lamp photographs of fluorescein staining in all treated eyes, and a protocol-driven method to quantify the outcomes.
The enrolled patients were randomized into one of two study groups, with patients receiving the same treatment in both eyes:
•Cohort 1 (n=117) was comprised of KIO-201 QID for two weeks after surgery.
•Cohort 2 (n=117) was comprised of BCL administered four times daily.
Study Results
KIO-201 demonstrated superiority for the primary endpoint with a p-value of 0.0203. The statistical significance measurement was based on the number of patients in each arm that achieved complete corneal defect closure three days post refractive surgery. At Day 3, 80.2% of eyes receiving the KIO-201 treatment regimen were completely healed, compared with 67.0% for BCL. Additionally, at Day 2, the average wound size for all eyes treated with KIO-201 was 3.61 mm2, compared to 6.66 mm2 for eyes treated with BCL, which is 46% smaller than the standard-of-care as noted in Figure 4. As described further, the use of KIO-201 resulted in smaller wounds in the
10
acute healing phase after PRK surgery compared to the standard-of-care (BCL). This data gives confidence that patients will be able to resume normal activities earlier when treated with KIO-201 compared to BCL.
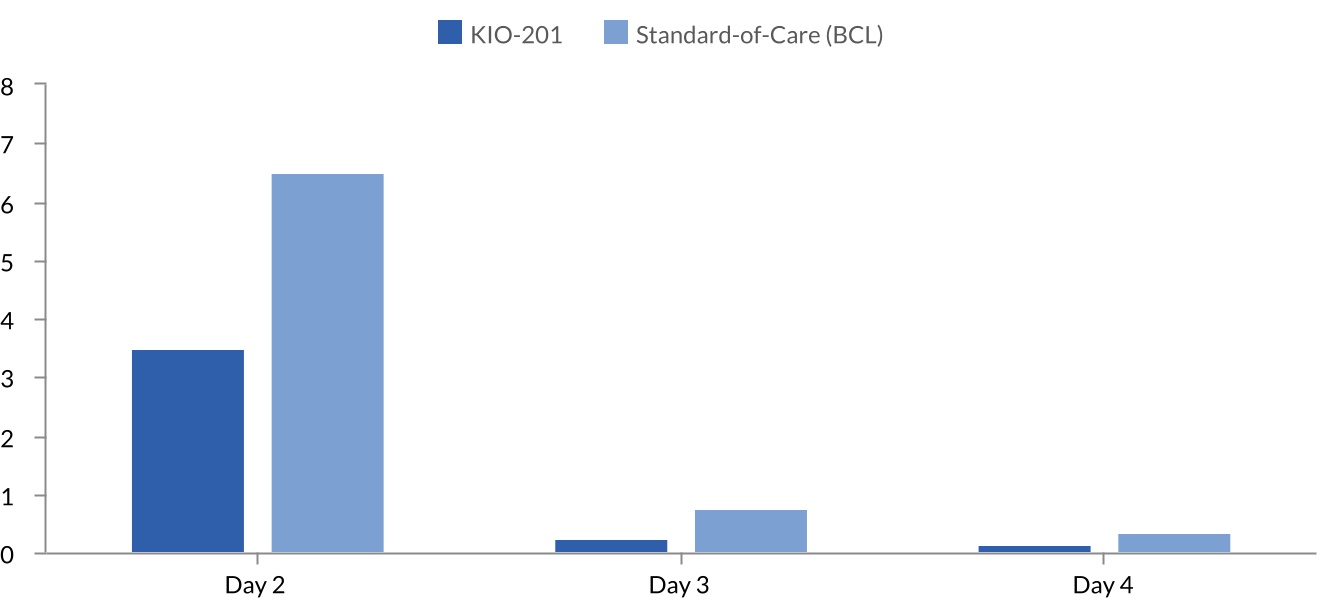
Figure 4: Mean wound size (mm2)
Clinical Development Plan
We are currently assessing the requirements on a registration clinical trial as well as evaluating the market opportunity. We expect to begin further clinical work in 2023.
KIO-301: Retinitis Pigmentosa
Phase 1b Study:
In Q4 2022, we initiated a clinical trial of KIO-301 in patients with later stage Retinitis Pigmentosa. We expect results from this study in late 2023.
Design
This is a Phase 1b open-label, single ascending dose clinical trial for people living with retinitis pigmentosa. The study will enroll six patients and evaluate 12 eyes. The first cohort of three patients will include individuals with no or bare light perception due to the progression of RP. The second cohort will include patients able to detect hand motion and count fingers. Dose escalations will be performed in each patient's contralateral eye. The primary endpoints are safety and tolerability, with secondary efficacy endpoints including objective and subjective evaluations, such as object identification and contrast assessment, navigation, perimetry, functional MRI and other ophthalmic and quality-of-life assessments. This multi-site study is being conducted at The Royal Adelaide Hospital (RAH) in Adelaide, South Australia as well as multiple private ophthalmology clinics in Adelaide, South Australia.
Clinical Development Plan
Retinitis pigmentosa is a rare inherited disease where patients typically present with loss of low light/night vision, followed by reduced visual field (loss of peripheral vision) and eventually loss of central vision. In Q1 2022, we received ODD from the FDA. We will assess the results from the Phase 1b clinical trial and determine next steps in the development of KIO-301 in late 2023.
11
Intellectual Property and Proprietary Rights
Overview
We are building an intellectual property portfolio for our KIO-101, KIO-201, and KIO-301 platforms and any other product candidates that we may develop, as well as other devices and product candidates for treatment of ocular indications in the U.S. and abroad. We currently seek, and intend to continue to seek, patent protection in the U.S. and internationally for our product candidates, methods of use, and processes for manufacture, and for other technologies, where appropriate. Our current policy is to actively seek to protect our proprietary position by, among other things, filing patent applications in the U.S. and abroad relating to proprietary technologies that are important to the development of our business. We also rely on, and will continue to rely on, trade secrets, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our technology.
Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection for the technologies that we consider important to our business, our ability to defend our patents, and our ability to preserve the confidentiality of our trade secrets and operate our business without infringing the patents and proprietary rights of third parties.
Patent Portfolio
Our patent portfolio includes patents covering KIO-101 including composition-of-matter, formulations thereof and its therapeutic uses in the treatment of ocular disorders and diseases and more. In addition, we hold a patent portfolio covering KIO-301 consisting of composition-of-matter, methods of use, and formulations thereof patents. Our KIO-201 portfolio of patents covers composition-of-matter and methods of use claims. These issued patents will expire between 2023 and 2036. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant new drug application or NDA. See “Government Regulation — Patent Term Restoration and Marketing Exclusivity” below.
Globally, we hold 26 active and valid patents.
License Agreements
We are a party to seven license agreements as described below. These license agreements require us to pay or receive royalties or fees to or from the licensor based on revenue or milestones related to the licensed technology.
On July 2, 2013, we (through our subsidiary, Kiora Pharmaceuticals, GmbH) entered into a patent and know-how assignment agreement with 4SC Discovery GmbH (4SC) transferring to us all patent rights and know-how to the compound KIO-101. We are responsible for paying royalties of 3.25% on net sales of KIO-101.
On July 2, 2013, we (through our subsidiary, Kiora Pharmaceuticals, GmbH) entered into an out-license agreement with 4SC granting 4SC the exclusive worldwide right to commercialize the compound KIO-101 for rheumatoid arthritis and inflammatory bowel disease, including Crohn’s disease and ulcerative colitis. We are eligible to receive milestone payments totaling up to €155 million, upon and subject to, the achievement of certain specified developmental and commercial milestones. We have not received any milestones from 4SC. In addition, we are eligible to receive royalties of 3.25% on net sales of KIO-101.
12
On September 12, 2013, we (through our subsidiary, Jade Therapeutics, Inc.) entered into an agreement with Lineage Cell Therapeutics, Inc. (Lineage), formerly known as BioTime, Inc., granting to us the exclusive worldwide right to commercialize cross-linked thiolated carboxymethyl hyaluronic acid (modified HA) for ophthalmic treatments in humans. The agreement requires us to pay an annual fee of $30,000 and a royalty of 6% on net sales of KIO-201 to Lineage based on revenue relating to any product incorporating the modified HA technology. The agreement expires when patent protection for the modified HA technology lapses in August 2027.
On November 17, 2014, we (through our subsidiary Kiora Pharmaceuticals GmbH) entered into an intellectual property and know-how licensing agreement with Laboratoires Leurquin Mediolanum S.A.S. (Mediolanum) for the commercialization of KIO-101 (the “Mediolanum Agreement”) in specific territories. Under the Mediolanum agreement, we out-licensed rights to commercialize KIO-101 for uveitis, dry eye, and viral conjunctivitis in Italy and France. This agreement was amended on December 10, 2015, to also include Belgium and The Netherlands. Under the Mediolanum Agreement, Mediolanum would be obligated to pay up to approximately €20 million in development and commercial milestones, and a 7% royalty on net sales of KIO-101 in the territories through the longer of the expiry of the valid patents covering KIO-101 or 10 years from the first commercial sale. The royalty would be reduced to 5% after patent expiry. During February 2023, we terminated the Mediolanum Agreement as permitted under its terms. Mediolanum has disputed the validity of the termination notice, and we are currently in communication with Mediolanum regarding the termination.
On September 26, 2018, we entered into an intellectual property licensing agreement (the “SentrX Agreement”) with SentrX, a veterinary medical device company that develops and manufactures veterinary wound care products. Under the SentrX Agreement, we in-licensed the rights to trade secrets and know-how related to the manufacturing of KIO-201. The SentrX Agreement enables us to pursue a different vendor with a larger capacity for manufacturing and an FDA-inspected facility for commercialization of a product for human use. Under the SentrX Agreement, SentrX is eligible to receive milestone payments totaling up to $4.75 million, upon and subject to the achievement of certain specified developmental and commercial milestones. The term of the agreement is until the Product is no longer in the commercial marketplace.
On May 1, 2020, we (through our subsidiary, Bayon Therapeutics, Inc.) entered into an agreement with the University of California (UC) granting to us the exclusive rights to its pipeline of photoswitch molecules. The agreement requires us to pay an annual fee to UC of $5,000, as well as payments to UC upon the achievement of certain development milestone and royalties based on revenue relating to any product incorporating KIO-301. The Company is obligated to pay royalties on net sales of 2% of the first $250 million of net sales, 1.25% of net sales between $250 million and $500 million, and 0.5% of net sales over $500 million. The agreement expires on the date of the last-to-expire patent included in the licensed patent portfolio which is January 2030.
On May 1, 2020, we (through our subsidiary, Bayon Therapeutics, Inc) entered into an agreement with Photoswitch Therapeutics, Inc. (Photoswitch) granting to us access to certain patent applications and IP rights with last-to-expire patent terms of January 2030. The agreement calls for payments to Photoswitch upon the achievement of certain development milestones and upon first commercial sale of the product.
Recent Developments
Reverse Stock Split
On September 23, 2022, we filed a Certificate of Amendment to our Restated Certificate of Incorporation (the “Amendment”) with the Secretary of State of the State of Delaware to effect a one-for-forty (1-for-40) reverse stock split of our outstanding common stock. The Amendment became effective at 12:01 a.m. Eastern Time on September 27, 2022. The Amendment was approved by our stockholders at our 2022 Annual Meeting of Stockholders held on September 23, 2022, and by our board of directors.
The Amendment provided that, at the effective time of the Amendment, every forty (40) shares of our issued and outstanding common stock automatically combined into one issued and outstanding share of common stock, without any change in par value per share. The reverse stock split affected all shares of our common stock outstanding immediately prior to the effective time of the Amendment. As a result of the reverse stock split, proportionate adjustments were made to the per share exercise price and/or the number of shares issuable upon the
13
exercise or vesting of all stock options, and restricted stock awards issued by us and outstanding immediately prior to the effective time of the Amendment, which resulted in a proportionate decrease in the number of shares of our common stock reserved for issuance upon exercise or vesting of such stock options, and restricted stock awards, and, in the case of stock options, a proportionate increase in the exercise price of all such stock options. In addition, the number of shares reserved for issuance under our equity compensation plans immediately prior to the effective time of the Amendment was reduced proportionately. The reverse stock split did not affect the number of shares or par value of common stock authorized for issuance under our Restated Certificate of Incorporation, which remained at 50,000,000 shares.
All share and per share amounts in this prospectus have been adjusted retroactively to reflect the reverse stock split as if it had occurred at the beginning of the earliest period presented. Our common stock began trading on The Nasdaq Capital Market on a split-adjusted basis when the market opened on September 27, 2022.
Warrant Inducement Transaction
On November 17, 2022, we entered into warrant exercise inducement offer letters (“Inducement Letters”) with certain of the investors from our July 2022 public offering, pursuant to which such investors agreed to exercise for cash all of their Class A Warrants to purchase 654,609 shares of our common stock originally issued in the Public Offering in exchange for our agreement to issue new warrants (the “Inducement Warrants”) on substantially the same terms as the Class A Warrants, except as described below, to purchase up to 654,609 shares of common stock (such issuance, the “Private Placement”). Each Inducement Warrant is exercisable at a price per share of common stock of $5.97. Each Inducement Warrant will initially be exercisable six months following its date of issuance, and will expire on the 18 month anniversary of their initial exercise date. We received aggregate gross proceeds of approximately $3.12 million from the exercise of the Class A Warrants by the selling stockholders and the sale of the Inducement Warrants.
Private Placement and Committed Equity Financing
On February 2, 2023, we entered into a securities purchase agreement (the “Securities Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“Lincoln Park”), pursuant to which we issued and sold to Lincoln Park (i) 52,798 shares of common stock and (ii) warrants to purchase an aggregate of up to 105,596 shares of common stock at an exercise price of $3.538 per share. The combined purchase price for each share of common stock and two warrants was $3.788. The private placement closed on February 3, 2023.
On February 2, 2023, and in connection with the Securities Purchase Agreement, we entered into a customary registration rights agreement with Lincoln Park, which requires us to, at the election of Lincoln Park, register the shares issued in the private placement on any registration statement that we file during the six-month period following the closing, subject to certain exceptions. However, the registration rights agreement does not afford any registration rights with respect to the warrants that we issued and sold to Lincoln Park in the private placement, or with respect to the underlying shares of common stock.
On February 3, 2023, we entered into a purchase agreement and a registration rights agreement with Lincoln Park. Under the terms and subject to the conditions of the purchase agreement, we have the right, but not the obligation, to sell to Lincoln Park, and Lincoln Park is obligated to purchase up to $10.0 million of our common stock. Such sales of common stock by us, if any, will be subject to certain limitations set forth in the purchase agreement, and may occur from time to time, at our sole discretion, over the 36-month period commencing on the date that the conditions to Lincoln Park’s purchase obligation set forth in the purchase agreement are satisfied, including that a registration statement covering the resale by Lincoln Park of shares of common stock that have been and may be issued to Lincoln Park under the Purchase Agreement, is declared effective by the SEC and a final prospectus relating thereto is filed with the SEC, which occurred on February 13, 2023 (the “Commencement Date”).
14
From and after the Commencement Date, we may from time to time on any business day, by written notice delivered by us to Lincoln Park, direct Lincoln Park to purchase up to 35,000 shares of common stock on such business day, at a purchase price per share that will be determined and fixed in accordance with the purchase agreement at the time we deliver such written notice to Lincoln Park (each, a “Regular Purchase”). The maximum number of shares we may sell to Lincoln Park in a Regular Purchase may be increased to up to 50,000 shares, with the applicable maximum share limit determined by whether the closing price for common stock on the applicable purchase date exceeds certain price thresholds set forth in the purchase agreement, in each case, subject to adjustment for any reorganization, recapitalization, non-cash dividend, stock split, reverse stock split or other similar transaction as provided in the purchase agreement. Lincoln Park’s maximum purchase commitment in any single Regular Purchase may not exceed $1,000,000. The purchase price per share of common stock sold in each such Regular Purchase, if any, will be based on prevailing market prices of the common stock immediately preceding the time of sale as computed under the Purchase Agreement.
In addition to Regular Purchases, provided that we have directed Lincoln Park to purchase the maximum amount of shares that we are then able to sell to Lincoln Park in a Regular Purchase and the closing sale price of the common stock is not below $1.00 per share on such date, we may, in our sole discretion, also direct Lincoln Park to purchase additional shares of common stock in “accelerated purchases,” and “additional accelerated purchases” as set forth in the purchase agreement. The purchase price per share of common stock sold in each such accelerated purchase and additional accelerated purchase, if any, will be based on prevailing market prices of the common stock at the time of sale as computed under the purchase agreement. There are no upper limits on the price per share that Lincoln Park must pay for shares of common stock in any purchase under the purchase agreement.
Our Corporate Information
Kiora Pharmaceuticals, Inc. was formed in Delaware on December 26, 2004 under the name EyeGate Pharmaceuticals, Inc. On November 8, 2021, we completed a merger of our wholly owned Delaware subsidiary, Kiora Pharmaceuticals, Inc. (incorporated in October 2021) into EyeGate Pharmaceuticals, Inc., which merger resulted in the amendment of our restated certificate of incorporation to change our name to “Kiora Pharmaceuticals, Inc.” effective November 8, 2021 (the “Name Change”). In connection with the name change, we changed our symbol on the Nasdaq Capital Market to “KPRX” and began using a new CUSIP number for shares of our common stock (49721T101) effective at the market open on November 8, 2021.We were originally incorporated in 1998 under the name of Optis France S.A. in Paris, France. We have four wholly owned subsidiaries: Jade Therapeutics, Inc., Kiora Pharmaceuticals, GmbH (formerly known as Panoptes Pharma Ges.m.b.H), Bayon Therapeutics, Inc., and Kiora Pharmaceuticals Pty Ltd (formerly known as Bayon Therapeutics Pty Ltd). Our former subsidiary, EyeGate Pharma S.A.S. was dissolved effective December 31, 2020. Our principal executive offices are located at 332 Encinitas Boulevard, Suite 102, Encinitas, California 92024, and our telephone number is (858) 224-9600. Our website address is www.kiorapharma.com. Our website and the information contained in, or accessible through, our website will not be deemed to be incorporated by reference into this prospectus and does not constitute part of this prospectus. You should not rely on any such information in making your decision whether to purchase our securities.
15
THE OFFERING
Securities offered by us |
2,582,160 shares of our common stock.
5,500 shares of Series F Preferred Stock that are convertible into an aggregate of up to 2,582,160 shares of common stock, subject to certain adjustments. For each share of common stock underlying a share of Series F Preferred Stock that we sell, the number of shares of common stock that we are selling will be decreased on a one-for-one basis.
Class C Warrants to purchase up to 2,582,160 shares of our common stock.
Class D Warrants to purchase up to 2,582,160 shares of our Common Stock.
|
||||
| Warrants | The warrants will be exercisable at an initial exercise price of $ per share. The warrants will include a one-time reset of the exercise price to a price equal to the lesser of (i) the then exercise price and (ii) 90% of the five-day volume weighted average prices for the five (5) trading days immediately preceding the date that is sixty calendar days after issuance of the warrants. The warrants will be exercisable beginning on the date of issuance. The Class C Warrants will expire on the five year anniversary of the date of issuance and the Class D Warrants will expire on the one year anniversary of the date of issuance. This prospectus also relates to the offering of the shares of common stock issuable upon exercise of the warrants. |
||||
Series F Preferred Stock |
Each share of Series F Preferred Stock is convertible at any time at the holder’s option into a number of shares of common stock equal to $1,000 divided by the Conversion Price. Notwithstanding the foregoing, we shall not effect any conversion of Series F Preferred Stock, to the extent that, after giving effect to an attempted conversion, the holder of shares of Series F Preferred Stock (together with such holder’s affiliates, and any persons acting as a group together with such holder or any of such holder’s affiliates) would beneficially own a number of shares of our common stock in excess of 4.99% of the shares of our common stock then outstanding after giving effect to such exercise. For additional information, see “Description of The Securities We Are Offering—Series F Convertible Preferred Stock” on page 59 of this prospectus. | ||||
Common stock outstanding after this offering |
4,606,430 shares, assuming that we sell all securities offered pursuant to this prospectus and assuming conversion of all shares of Series F Preferred Stock but no exercise of the warrants issued in this offering. | ||||
Price per share of common stock |
$2.13 combined public offering price per share of common stock, Class C Warrant and Class D Warrant based upon an assumed combined public offering price of $2.13, the closing price of our common stock on May 30, 2023. | ||||
Price per share of Series F Preferred Stock and warrants |
$1,000 | ||||
16
Underwriters’ option to purchase additional shares |
We have granted the underwriters an option, exercisable for forty-five (45) days after the date of this prospectus, to purchase up to an additional 387,324 shares of common stock, Class C Warrants to purchase 387,324 shares of common stock and/or Class D Warrants to purchase 387,324 shares of common stock at the assumed combined public offering price per share of common stock and warrant set forth on the cover page hereto less the underwriting discounts and commission. |
||||
Use of proceeds |
We intend to use the net proceeds from this offering to support our operations, including for clinical trials, for working capital and for other general corporate purposes. See “Use of Proceeds” on page 55.
|
||||
Risk factors |
Investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 18 of this prospectus, and under similar headings in the other documents that are incorporated by reference into this prospectus, for a discussion of information that should be considered in connection with an investment in our securities. | ||||
| Nasdaq Capital Market symbol | KPRX. We do not plan on applying to list the Class C Warrants, Class D Warrants or the Series F Preferred Stock on Nasdaq, any national securities exchange or any other nationally recognized trading system. Without an active trading market, the liquidity of the Class C Warrants, Class D Warrants and the Series F Preferred Stock will be limited. |
||||
The number of shares of our common stock to be outstanding after this offering is based on 2,024,270 shares of our common stock outstanding as of May 30, 2023, assumes no exercise of the underwriters’ over-allotment option, and assumes that the shares of Series F Preferred Stock sold in the offering have been converted, but does not include, as of such date:
•202,126 shares of common stock issuable upon exercise of options outstanding under our 2005 and 2014 Equity Incentive Plans, at a weighted-average exercise price of approximately $17.43 per share;
•67,250 shares of common stock granted through a restricted stock award subject to release under our 2005 and 2014 Equity Incentive Plans;
•1,613,483 shares of our common stock issuable upon the exercise of outstanding warrants to purchase shares of our common stock with a weighted-average exercise price of $16.33 per share;
•23,927 shares of common stock reserved for future issuance under our 2014 Equity Incentive Plan;
•191 shares of common stock reserved for future issuance under our 2014 Employee Stock Purchase Plan;
•52 shares of common stock issuable upon the conversion of outstanding shares of Series D Convertible Preferred Stock;
•2,582,160 shares of common stock issuable upon the exercise of Class C Warrants to be issued to investors in this offering at an exercise price of $ per share; and
•2,582,160 shares of common stock issuable upon the exercise of Class D Warrants to be issued to investors in this offering at an initial exercise price of $ per share.
17
RISK FACTORS
Summary of Risk Factors
Below is a summary of the principal factors that make an investment in our securities speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face, can be found below under the heading “Risk Factors” and should be carefully considered, together with other information in this prospectus and the documents incorporated by reference herein before making an investment decision regarding our securities.
•We have incurred significant operating losses since our inception, which have caused management to determine there is substantial doubt regarding our ability to continue as a going concern. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
•We will need substantial additional funding. If we are unable to raise capital when needed, we could be forced to delay, reduce, or eliminate our product development programs or commercialization efforts.
•Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
•Our limited operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
•Our future success depends on our ability to retain key executives and to attract, retain, and motivate qualified personnel.
•The coronavirus pandemic could adversely impact our business, including clinical trials.
•We depend heavily on the success of KIO-101, KIO-201, and KIO-301. If we are unable to successfully obtain marketing approval for KIO-101, KIO-201, or KIO-301, or experience significant delays in doing so, or if after obtaining marketing approvals, we fail to commercialize KIO-101, KIO-201, or KIO-301, our business will be materially harmed.
•If clinical trials of KIO-101, KIO-201, KIO-301, or any other product candidate that we develop fail to demonstrate safety and efficacy to the satisfaction of the FDA or foreign regulatory authorities or do not otherwise produce favorable results, we may incur additional costs or experience delays in completing, or ultimately be delayed or unable to complete, the development and commercialization of KIO-101, KIO-201, KIO-301, or any other product candidate.
•Even if KIO-101, KIO-201, KIO-301, or any other product candidate that we develop receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success and the market opportunity for our product candidates may be smaller than we estimate.
•If we are unable to establish sales, marketing, and distribution capabilities, we may not be successful in KIO-101, KIO-201, KIO-301, or any other product candidates that we may develop if and when they are approved.
•We face substantial competition, which may result in others discovering, developing, or commercializing products before or more successfully than we do.
•Even if we are able to commercialize KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop, the products may become subject to unfavorable pricing regulations, third-party coverage or reimbursement practices, or healthcare reform initiatives which could harm our business.
•We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.
18
•If we are unable to obtain and maintain patent protection for our technology and products or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
•We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming, and unsuccessful.
•Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
•If we are not able to obtain required regulatory approvals, we will not be able to commercialize KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop; and our ability to generate revenue will be materially impaired.
•We incur increased costs as a result of operating as a public company, and our management is required to devote substantial time to new compliance initiatives and corporate governance practices.
•Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
•We have identified a material weakness in our internal control over financial reporting. If we are unable to remediate this material weakness, or if we experience additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls, we may not be able to accurately or timely requirements applicable to public companies, which may adversely affect investor confidence in us, and, as a result, the market price of our common stock.
Risk Factors
Investing in our securities involves a high degree of risk. You should carefully consider the risks and uncertainties and all other information contained in or incorporated by reference in this prospectus. All of these risk factors are incorporated by reference herein in their entirety. Our business, financial condition or results of operations could be materially adversely affected by the materialization of any of these risks. The trading price of our securities could decline due to the materialization of any of these risks, and you may lose all or part of your investment. This prospectus and the documents incorporated herein by reference also contain forward-looking statements that involve risks and uncertainties. Actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including the risks described herein and in the documents incorporated herein by reference.
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred significant operating losses since our inception, which have caused management to determine there is substantial doubt regarding our ability to continue as a going concern. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
Since inception, we have incurred significant operating losses. Our net loss was approximately $13.6 million for the year ended December 31, 2022, $13.8 million for the year ended December 31, 2021 and $134.5 million from the period of inception (December 26, 2004) through December 31, 2022. To date, we have financed our operations primarily through private placements and public offerings of our securities, and payments from our license agreements. We have devoted substantially all of our financial resources and efforts to research and development, including pre-clinical studies and, beginning in 2008, clinical trials. We are still in the development stage of our product candidates and we have not completed development of any drugs. We expect to continue to incur significant expenses and operating losses for the foreseeable future. Our net losses may fluctuate significantly from quarter to quarter and year to year. Our recurring losses from operations have caused management to determine there is substantial doubt regarding our ability to continue as a going concern, and as a result, our independent registered
19
public accounting firm included an explanatory paragraph in its report on our consolidated financial statements as of and for the year ended December 31, 2022, with respect to this uncertainty.
We anticipate that our expenses will continue to be significant with the clinical trials for the ongoing development of our KIO-101, KIO-201, and KIO-301 products.
Our expenses will also increase if and as we:
•seek marketing approval for KIO-101, KIO-201, and KIO-301, whether alone or in collaboration with third parties;
•continue the research and development of KIO-101, KIO-201, KIO-301, and any of our other product candidates;
•seek to develop additional product candidates;
•in-license or acquire the rights to other products, product candidates, or technologies;
•seek marketing approvals for any product candidates that successfully complete clinical trials;
•establish sales, marketing, and distribution capabilities and scale up and validate external manufacturing capabilities to commercialize any products for which we may obtain marketing approval;
•maintain, expand and protect our intellectual property portfolio;
•hire additional clinical, quality control, scientific and management personnel;
•expand our operational, financial and management systems, and personnel, including personnel to support our clinical development, manufacturing, and planned future commercialization efforts and our operations as a public company; and
•increase our insurance coverage as we expand our clinical trials and commence commercialization of KIO-101, KIO-201, and KIO-301.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. Our expenses will increase if:
•we are required by the FDA or foreign equivalents to perform studies or clinical trials in addition to those currently expected; and
•there are any delays in enrollment of patients in or completing our clinical trials or the development of KIO-101, KIO-201, KIO-301, or any other product candidates that we may develop.
Our ability to become and remain profitable depends on our ability to generate revenue. We do not expect to generate significant revenue unless and until we obtain marketing approval for, and commercialize KIO-101, KIO-201, KIO-301, or other product candidates that we may develop, which may never occur. This will require us to be successful in a range of challenging activities, including:
•establishing collaboration, distribution, or other marketing arrangements with third parties to commercialize KIO-101, KIO-201, and KIO-301 in markets outside the U.S.;
•achieving an adequate level of market acceptance of our product candidates;
•protecting our rights to our intellectual property portfolio related to our product candidates; and
•ensuring the manufacture of commercial quantities of KIO-101, KIO-201, and KIO-301.
20
Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings, or even continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
We will need substantial additional funding. If we are unable to raise capital when needed, we could be forced to delay, reduce, or eliminate our product development programs or commercialization efforts.
We expect to devote substantial financial resources to our ongoing and planned activities, particularly continuing the clinical development of our KIO-101, KIO-201, and KIO-301 products. In the future, we expect to raise additional financial resources for the continued clinical development of KIO-101, KIO-201, KIO-301, and other product candidates we may develop. In addition, if we obtain regulatory approval for any of our product candidates, we would need to devote substantial financial resources to commercialization efforts, including product manufacturing, marketing, sales, and distribution. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce, or eliminate our research and development programs or any future commercialization efforts.
Our future capital requirements will depend on many factors, including:
•the progress, costs, and outcome of our clinical trials for our product candidates and of any clinical activities required for regulatory review of our product candidates outside of the U.S.;
•the costs and timing of process development and manufacturing scale up and validation activities associated with our product candidates;
•the costs, timing, and outcome of regulatory review of our product candidates in the U.S., and in other jurisdictions;
•the costs and timing of commercialization activities for our product candidates if we receive marketing approval, including the costs and timing of establishing product sales, marketing, distribution, and outsourced manufacturing capabilities;
•subject to receipt of marketing approval, the amount of revenue received from commercial sales of our product candidates;
•our ability to establish collaborations on favorable terms, if at all, particularly manufacturing, marketing, and distribution arrangements for our product candidates;
•the costs and timing of preparing, filing, and prosecuting patent applications, maintaining and enforcing our intellectual property rights, and defending any intellectual property-related claims; and
•the extent to which we in-license or acquire rights to other products, product candidates, or technologies for the treatment of ophthalmic diseases.
As of March 31, 2023, we had cash and cash equivalents of $3.4 million. We believe we will have sufficient cash to fund planned operations into July 2023, however, the acceleration or reduction of cash outflows by management can significantly impact the timing needed for raising additional capital to complete development of our products. To continue development, we will need to raise additional capital through debt and/or equity financing or access additional funding through U.S. or foreign grants. Although we completed our initial public offering and subsequent public offerings, registered direct offerings and private placements, additional capital may not be available on terms favorable to us, if at all. Accordingly, no assurances can be given that management will be successful in these endeavors. These conditions raise substantial doubt about our ability to continue as a going concern. The consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities or any other adjustments that might be necessary should we be unable to continue as a going concern.
21
Identifying potential product candidates and conducting pre-clinical testing and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain regulatory approval and achieve product sales. Our commercial revenues, if any, will be derived from sales of KIO-101, KIO-201, KIO-301, or any other products that we successfully develop, none of which we expect to be commercially available for several years, if at all. In addition, if approved, any product candidate that we develop or any product that we in-license may not achieve commercial success. Accordingly, we will need to obtain substantial additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings, government or other third-party funding, collaborations, strategic alliances, licensing arrangements, and marketing and distribution arrangements. We do not have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we cannot raise funds on acceptable terms, we may not be able to grow our business or respond to competitive pressures.
If we raise additional funds through government or other third-party funding, collaborations, strategic alliances, licensing arrangements, or marketing and distribution arrangements, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce, or terminate our product development or future commercialization efforts, or grant rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves.
Our limited operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We are a clinical-stage company with a limited operating history. Our operations to date have been limited to organizing and staffing our company, acquiring rights to intellectual property, business planning, raising capital, developing our technology, identifying potential product candidates, undertaking preclinical studies, and conducting clinical trials of KIO-101, KIO-201, and KIO-301. We have not yet demonstrated our ability to successfully complete development of a product candidate, obtain marketing approvals, manufacture at commercial scale, or arrange for a third party to do so on our behalf, or conduct sales, marketing, and distribution activities necessary for successful product commercialization.
In addition, as a pre-revenue business, we may encounter unforeseen expenses, difficulties, complications, delays, and other known and unknown factors. We will need to transition at some point from a company with a research and development focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
We expect our financial condition and operating results to continue to fluctuate significantly from quarter-to-quarter and year-to-year due to a variety of factors, many of which are beyond our control. Accordingly, you should not rely upon the results of any quarterly or annual periods as indications of future operating performance.
22
Foreign currency exchange rate fluctuations may have a negative impact on our financial results.
We are subject to the risks of fluctuating foreign currency exchange rates, which could have an adverse effect on the costs and expenses of our foreign subsidiaries. As a result, currency fluctuations among the U.S. dollar, euro, Australian dollar, and the other currencies in which we do business have caused and will continue to cause foreign currency translation and transaction gains and losses. We have not used forward exchange contracts to hedge our foreign currency exposures. In the future, we may undertake to manage foreign currency risk through hedging methods, including foreign currency contracts. We recognize foreign currency gains or losses arising from our operations in the period incurred. We cannot guarantee that we will be successful in managing foreign currency risk or in predicting the effects of exchange rate fluctuations upon our future operating results because of the number of currencies involved, the variability of currency exposure, and the potential volatility of currency exchange rates. We cannot predict with any certainty changes in foreign currency exchange rates or the degree to which we can address these risks.
Risks Related to the Discovery and Development of Our Product Candidates
We depend heavily on the success of KIO-101, KIO-201, and KIO-301. If we are unable to successfully obtain marketing approval for KIO-101, KIO-201, and KIO-301, or experience significant delays in doing so, or if after obtaining marketing approvals, we fail to commercialize KIO-101, KIO-201, and KIO-301, our business will be materially harmed.
We have invested a significant portion of our efforts and financial resources in the development of KIO-201, and we expect to invest a significant portion of our efforts and financial resources in the development of KIO-101 and KIO-301 in the future. There remains a significant risk that we will fail to successfully develop either product candidate.
We cannot accurately predict when or if KIO-101, KIO-201, or KIO-301 will prove effective or safe in humans or whether it will receive marketing approval. Our ability to generate product revenues, which may never occur, will depend heavily on our obtaining marketing approval for and commercializing KIO-101, KIO-201, and KIO-301.
The success of KIO-101, KIO-201, and KIO-301 will depend on several factors, including the following:
•obtaining favorable results from clinical trials;
•applying for and receiving marketing approvals from applicable regulatory authorities for KIO-101, KIO-201, and KIO-301;
•making arrangements with third-party manufacturers for commercial quantities of KIO-101, KIO-201, and KIO-301 and receiving regulatory approval of our manufacturing processes and our third-party manufacturers’ facilities from applicable regulatory authorities;
•establishing sales, marketing, and distribution capabilities and launching commercial sales of KIO-101, KIO-201, and KIO-301, if and when approved, whether alone or in collaboration with others;
•acceptance of KIO-101, KIO-201, and KIO-301, if and when approved, by patients, the medical community, and third-party payors;
•effectively competing with other therapies, including the existing standard-of-care;
•maintaining a continued acceptable safety profile of KIO-101, KIO-201, and KIO-301 following approval;
•obtaining and maintaining coverage and adequate reimbursement from third-party payors;
•obtaining and maintaining patent and trade secret protection and regulatory exclusivity; and
•protecting our rights in our intellectual property portfolio related to KIO-101, KIO-201, and KIO-301.
23
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize KIO-101, KIO-201, and KIO-301, which would materially harm our business.
If clinical trials of KIO-101, KIO-201, KIO-301, or any other product candidate that we develop fail to demonstrate safety and efficacy to the satisfaction of the FDA or foreign regulatory authorities or do not otherwise produce favorable results, we may incur additional costs or experience delays in completing, or ultimately be delayed or unable to complete, the development and commercialization of KIO-101, KIO-201, KIO-301, or any other product candidate.
Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete pre-clinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Clinical testing is expensive, difficult to design and implement, can take many years to complete, and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. The outcome of preclinical testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, pre-clinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products.
If we experience any of a number of possible unforeseen events in connection with our clinical trials, potential marketing approval or commercialization of our product candidates could be delayed or prevented.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing approval or commercialize KIO-101, KIO-201, KIO-301, or any other product candidates that we may develop, including:
•clinical trials of our product candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs;
•the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate, or participants may drop out of these clinical trials at a higher rate than we anticipate;
•any third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all;
•regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site;
•we may experience delays in reaching, or fail to reach, agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites;
•we may decide, or regulators or institutional review boards may require us, to suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks;
•the cost of clinical trials of our product candidates may be greater than we anticipate;
•the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; and
•our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators, or institutional review boards to suspend or terminate the trials.
24
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not favorable or are only modestly favorable or if there are safety concerns, we may:
•be delayed in obtaining marketing approval for our product candidates;
•not obtain marketing approval at all;
•obtain approval for indications or patient populations that are not as broad as intended or desired;
•obtain approval with labeling that includes significant use or distribution restrictions or safety warnings;
•be subject to additional post-marketing testing requirements; or
•have the product removed from the market after obtaining marketing approval.
Our product development costs will also increase if we experience delays in testing or marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, or at all. Significant pre-clinical or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates.
If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for KIO-101, KIO-201, and KIO-301, or our other product candidates that we may develop if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the U.S. In addition, some of our competitors may have ongoing clinical trials for product candidates that treat the same indications as KIO-101, KIO-201, and KIO-301, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ product candidates.
Our inability to enroll a sufficient number of patients for our clinical trials would result in significant delays, could require us to abandon one or more clinical trials altogether, and could delay or prevent our receipt of necessary regulatory approvals. Enrollment delays in our clinical trials may result in increased development costs for our product candidates, which would cause the value of our company to decline and limit our ability to obtain additional financing.
If serious adverse or unacceptable side effects are identified during the development of our product candidates, we may need to abandon or limit our development of such product candidates.
If KIO-101, KIO-201, KIO-301, or any of our other product candidates are associated with serious adverse events or undesirable side effects in clinical trials or have characteristics that are unexpected, we may need to abandon their development or limit development to more narrow uses or subpopulations in which the serious adverse events, undesirable side effects, or other characteristics are less prevalent, less severe, or more acceptable from a risk-benefit perspective. Many compounds that initially showed promise in clinical or early-stage testing for treating ophthalmic disease have later been found to cause side effects that prevented further development of the compound.
25
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs and product candidates that we identify for specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing, or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. To the extent our contemplated trials are unsuccessful, we may not be able to raise additional funds for subsequent trials or pursuing other indications.
Risks Related to the Commercialization of Our Product Candidates
Even if KIO-101, KIO-201, KIO-301, or any other product candidate that we develop receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors, and others in the medical community necessary for commercial success and the market opportunity for our product candidates may be smaller than we estimate.
If KIO-101, KIO-201, KIO-301, or any other product candidate that we develop receives marketing approval, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors, and others in the medical community.
Our assessment of the potential market opportunity for KIO-101, KIO-201, and KIO-301 is based on industry and market data that we obtained from industry publications and research, surveys, and studies conducted by third parties. Industry publications and third-party research, surveys, and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. If the actual market for KIO-101, KIO-201, and KIO-301 is smaller than we expect, our product revenue may be limited, and it may be more difficult for us to achieve or maintain profitability.
If we are unable to establish sales, marketing and distribution capabilities, we may not be successful in KIO-101, KIO-201, KIO-301, or any other product candidates that we may develop if and when they are approved.
We do not have a sales or marketing infrastructure. To achieve commercial success for any product for which we have obtained marketing approval and have not licensed the commercialization rights, we will need to establish sales, marketing, and distribution capabilities, either ourselves or through collaborations or other arrangements with third parties.
In the future, we plan to build sales and marketing infrastructure to market or co-promote KIO-101, KIO-201, KIO-301, and possibly other product candidates that we develop, if and when they are approved. There are risks involved with establishing our own sales, marketing, and distribution capabilities. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of KIO-101, KIO-201, KIO-301, or any other product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize product candidates on our own include:
•our inability to recruit, train, and retain adequate numbers of effective sales and marketing personnel;
26
•the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe our products;
•the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
•unforeseen costs and expenses associated with creating an independent sales and marketing organization.
We expect to enter into arrangements with third parties to perform consulting, sales, marketing, and distribution services in markets outside the U.S. We may also enter into arrangements with third parties to perform these services in the U.S. if we do not establish our own sales, marketing, and distribution capabilities in the U.S., or if we determine that such third-party arrangements are otherwise beneficial. Our product revenues and our profitability, if any, under any such third-party sales, marketing, or distribution arrangements are likely to be lower than if we were to market, sell, and distribute our product candidates. In addition, we may not be successful in entering into arrangements with third parties to sell, market, and distribute our product candidates, or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our product candidates effectively. If we do not establish sales, marketing, and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing KIO-101, KIO-201, KIO-301 or any other product candidates that we may develop.
We face substantial competition, which may result in others discovering, developing, or commercializing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to KIO-101, KIO-201, KIO-301, and our other current product candidates and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies worldwide. There are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of products for the treatment of the disease indications for which we are developing our product candidates. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent protection, and establish collaborative arrangements for research, development, manufacturing, and commercialization.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient, or are less expensive than our product candidates. Our competitors also may obtain FDA or foreign regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
In addition, our ability to compete may be affected in many cases by insurers or other third-party payors, particularly Medicare, seeking to encourage the use of generic products. Generic products are currently being used for the indications that we are pursuing, and additional products are expected to become available on a generic basis over the coming years. If KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop achieves marketing approval, we expect that it will be priced at a premium over competitive products.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites, and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
27
Even if we are able to commercialize KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop, the products may become subject to unfavorable pricing regulations, third-party coverage or reimbursement practices, or healthcare reform initiatives, which could harm our business.
Our ability to commercialize KIO-101, KIO-201, KIO-301, or any other product candidates that we may develop successfully will depend, in part, on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government healthcare programs, private health insurers, managed care plans, and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Coverage and reimbursement may not be available for our product candidates and, even if they are available, the level of reimbursement may not be satisfactory.
Inadequate reimbursement may adversely affect the demand for, or the price of, any product candidate for which we obtain marketing approval. Obtaining and maintaining adequate reimbursement for our products may be difficult. We may be required to conduct expensive pharmacoeconomic studies to justify coverage and reimbursement or the level of reimbursement relative to other therapies. If coverage and adequate reimbursement are not available or reimbursement is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the indications for which the drug is approved by the FDA or similar regulatory authorities outside the U.S. Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale, and distribution expenses. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the U.S. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that we develop would compromise our ability to generate revenues and become profitable.
The regulations that govern marketing approvals, pricing, coverage, and reimbursement for new drug products vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenues we are able to generate from the sale of the product in that country. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval.
There can be no assurance that our product candidates or any products that we may in-license, if they are approved for sale in the U.S. or in other countries, will be considered medically reasonable and necessary for a specific indication, that they will be considered cost-effective by third-party payors, that coverage and an adequate
28
level of reimbursement will be available, or that third-party payors’ reimbursement policies will not adversely affect our ability to sell our product candidates profitably.
Our strategy of obtaining rights to product candidates and approved products through in-licenses and acquisitions may not be successful.
We may expand our product pipeline through opportunistically in-licensing or acquiring the rights to other products, product candidates, or technologies. The future growth of our business may depend in part on our ability to in-license or acquire the rights to approved products, additional product candidates, or technologies. However, we may be unable to in-license or acquire the rights to any such products, product candidates, or technologies from third parties. The in-licensing and acquisition of pharmaceutical products is a competitive area, and a number of more established companies are also pursuing strategies to license or acquire products, product candidates, or technologies that we may consider attractive. These established companies may have a competitive advantage over us due to their size, cash resources, and greater clinical development and commercialization capabilities.
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to in-license or acquire the rights to the relevant product, product candidate, or technology on terms that would allow us to make an appropriate return on our investment. Furthermore, we may be unable to identify suitable products, product candidates, or technologies within our area of focus. If we are unable to successfully obtain rights to suitable products, product candidates or technologies, our ability to pursue this element of our strategy could be impaired.
Product liability lawsuits against us could cause us to incur substantial liabilities and limit commercialization of any products that we develop.
We face an inherent risk of product liability exposure related to the use of the product candidates that we develop in human clinical trials and will face an even greater risk if we commercially sell any products that we develop. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•decreased demand for any product candidates or products that we develop;
•injury to our reputation and significant negative media attention;
•withdrawal of clinical trial participants;
•significant costs to defend the related litigation;
•substantial monetary awards to trial participants or patients;
•loss of revenue;
•reduced time and attention of our management to pursue our business strategy; and
•the inability to commercialize any products that we develop.
While we obtain insurance for each clinical trial we perform, we may not be adequately insured to cover all liabilities that we may incur. We will need to increase our insurance coverage as we expand our clinical trials. We will need to further increase our insurance coverage if we commence commercialization of any product candidate that receives marketing approval. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
29
Risks Related to Our Dependence on Third Parties
We may enter into collaborations with other third parties for the development or commercialization of our product candidates. If our collaborations are not successful, we may not be able to capitalize on the market potential of these product candidates.
We expect to utilize a variety of types of collaboration, distribution, and other marketing arrangements with third parties to commercialize KIO-101, KIO-201, and KIO-301 in markets outside the U.S. We also may enter into arrangements with third parties to perform these services in the U.S. if we do not establish our own sales, marketing, and distribution capabilities in the U.S., or if we determine that such third-party arrangements are otherwise beneficial. We also may seek third-party collaborators for development and commercialization of other product candidates. Our likely collaborators for any sales, marketing, distribution, development, licensing, or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, and biotechnology companies. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities and efforts to successfully perform the functions assigned to them in these arrangements.
Collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner, or at all. If we do not receive the funding we expect under any future collaboration agreements, our development of our product candidates could be delayed and we may need additional resources to develop our product candidates. All of the risks relating to product development, regulatory approval, and commercialization described in this prospectus also apply to the activities of our collaborators.
Additionally, subject to its contractual obligations to us, if a collaborator of ours were to be involved in a business combination, it might de-emphasize or terminate the development or commercialization of any product candidate licensed to it by us. If one of our collaborators terminates its agreement with us, we may find it more difficult to attract new collaborators and our perception in the business and financial communities could be harmed.
If we are not able to establish additional collaborations, we may have to alter our development and commercialization plans and our business could be adversely affected.
For some of our product candidates, we may decide to collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of therapeutic products. We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration, and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the U.S., the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. We may also be restricted under future license agreements from entering into agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
If we are unable to reach agreements with suitable collaborators on a timely basis, on acceptable terms, or at all, we may have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to fund and undertake development or commercialization activities on our own, we may need to obtain additional expertise and additional capital, which may not be available to us on acceptable terms or at all. If we fail to enter into collaborations and do not have sufficient funds or expertise to undertake the necessary
30
development and commercialization activities, we may not be able to further develop our product candidates or bring them to market or continue to develop our product platform.
We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.
We have relied on third parties, such as contract research organizations (CROs) to conduct our completed trials of our product candidates, and do not plan to independently conduct clinical trials of our product candidates. We expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions, and clinical investigators, to conduct our clinical trials. These agreements might terminate for a variety of reasons, including a failure to perform by the third parties. If we need to enter into alternative arrangements, that would delay our product development activities.
Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical Practices, for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible and accurate, and that the rights, integrity and confidentiality of trial participants are protected. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity, and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
We contract with third parties for the manufacture of KIO-101, KIO-201, and KIO-301 for clinical trials and expect to continue to do so in connection with the commercialization of KIO-101, KIO-201, KIO-301, and for clinical trials and commercialization of any other product candidates that we may develop. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or products or such quantities at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of KIO-101, KIO-201, KIO-301, or any other of our product candidates. We rely, and expect to continue to rely, on third parties to manufacture clinical and commercial supplies of KIO-101, KIO-201, and KIO-301, pre-clinical and clinical supplies of our other product candidates that we may develop, and commercial supplies of products if and when any of our product candidates receive marketing approval. Our current and anticipated future dependence upon others for the manufacture of KIO-101, KIO-201, KIO-301, and any other product candidate or product that we develop, may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis. In addition, any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval.
We currently rely exclusively on third-party manufacturers to assemble and prepare KIO-101, KIO-201, and KIO-301 on a purchase order basis. We do not currently have any contractual commitments for commercial supply of bulk drug substance for KIO-101, KIO-201, KIO-301, or fill-finish services. We also do not currently have arrangements in place for redundant supply or a second source for bulk drug substance for KIO-101, KIO-201, and KIO-301, or for fill-finish services. The prices at which we are able to obtain supplies of KIO-101, KIO-201, KIO-301, and fill-finish services may vary substantially over time and adversely affect our financial results.
If our third-party manufacturers for KIO-101, KIO-201, or KIO-301 fail to fulfill our purchase orders or should become unavailable to us for any reason, we believe that there are a limited number of potential replacement manufacturers, and we likely would incur added costs and delays in identifying or qualifying such replacements. We
31
could also incur additional costs and delays in identifying or qualifying a replacement manufacturer for fill-finish services if our existing third-party manufacturer should become unavailable for any reason. We may be unable to establish any agreements with such replacement manufacturers or to do so on acceptable terms. Even if we could transfer manufacturing to a different third party, the shift would likely be expensive and time consuming, particularly since the new facility would need to comply with the necessary regulatory requirements and we would need FDA approval before using or selling any products manufactured at that facility.
In connection with our application for a license to market KIO-101, KIO-201, KIO-301, or other product candidates in the U.S., we may be required to conduct a comparability study if the product we intend to market is supplied by a manufacturer different from the one who supplied the product evaluated in our clinical studies. Delays in designing and completing this study to the satisfaction of the FDA could delay or preclude our development and commercialization plans and thereby limit our revenues and growth.
Reliance on third-party manufacturers entails additional risks, including:
•KIO-101, KIO-201, KIO-301, and any other product candidates that we may develop may compete with other product candidates and products for access to a limited number of suitable manufacturing facilities that operate under current good manufacturing practices or CGMP regulations;
•reliance on the third party for regulatory compliance and quality assurance;
•the possible breach of the manufacturing agreement by the third party;
•the possible misappropriation of our proprietary information, including our trade secrets and know-how; and
•the possible termination or non-renewal of the agreement by the third party at a time that is costly or inconvenient for us.
Third-party manufacturers may not be able to comply with CGMP regulations or similar regulatory requirements outside the U.S. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions, and criminal prosecutions, any of which could significantly and adversely affect supplies of our products.
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain patent protection for our technology and products or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
Our success depends in large part on our ability to obtain and maintain patent protection in the U.S. and other countries with respect to our proprietary technology and products. We and our licensors have sought to protect our proprietary position by filing patent applications in the U.S. and abroad related to our novel technologies and product candidates. The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Maintaining patents in the U.S. is an expensive process and it is even more expensive to maintain patents and patent applications in foreign countries. As a result, it is possible that we and our licensors will fail to maintain such patents thereby reducing the rights of our portfolio.
The patent position of pharmaceutical, biotechnology, and medical device companies generally is highly uncertain, involves complex legal and factual questions, and has in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability, and commercial value of our and our licensors’ patent rights are highly uncertain. Our and our licensors’ pending and future patent applications may not result in patents being
32
issued which protect our technology or products, or which effectively prevent others from commercializing competitive technologies and products. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the U.S. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the U.S. and other jurisdictions are typically not published until 18 months after filing, or in some cases, not at all. Therefore, we cannot know with certainty whether we or our licensors were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability, and commercial value of our owned or licensed patent rights are highly uncertain. We currently have 57 pending patents. Our pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. In particular, during prosecution of any patent application, the issuance of any patents based on the application may depend upon our ability to generate additional pre-clinical or clinical data that support the patentability of our proposed claims. We may not be able to generate sufficient additional data on a timely basis, or at all. Moreover, changes in either the patent laws or interpretation of the patent laws in the U.S. and other countries may diminish the value of our patents or narrow the scope of our patent protection.
Moreover, we may be subject to a third-party pre-issuance submission of prior art to the U.S. Patent and Trademark Office, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review, or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding, or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop, or commercialize current or future product candidates.
Even if our owned and licensed patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us, or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity, or enforceability and our owned and licensed patents may be challenged in the courts or patent offices in the U.S. and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated, or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing, and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming, and unsuccessful.
Competitors may infringe our issued patents or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringed their patents. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly, or refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
33
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability, and the ability of our collaborators, to develop, manufacture, market, and sell our product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. There is considerable intellectual property litigation in the medical device, biotechnology, and pharmaceutical industries. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology, including interference or derivation proceedings before the U.S. Patent and Trademark Office. The risks of being involved in such litigation and proceedings may increase as our product candidates near commercialization and as we gain the greater visibility associated with being a public company. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future. We may not be aware of all such intellectual property rights potentially relating to our product candidates and their uses. Thus, we do not know with certainty that KIO-101, KIO-201, KIO-301, or any other product candidate, or our commercialization thereof, does not and will not infringe or otherwise violate any third party’s intellectual property.
If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our products and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
If we fail to comply with our obligations in our intellectual property licenses and funding arrangements with third parties, we could lose rights that are important to our business.
We are party to licensing agreements that impose, and for a variety of purposes, we will likely enter into additional licensing and funding arrangements with third parties that may impose, diligence, development, and commercialization timelines and milestone payment, royalty, insurance, and other obligations on us. Under certain of our existing licensing agreements, we are obligated to pay royalties or make specified milestone payments on net product sales to the extent they are covered by the agreements. We also are obligated under certain of our existing license agreements to pay maintenance and other fees. We also have diligence and development obligations under certain of those agreements that we are required to satisfy. If we fail to comply with our obligations under current or future license and collaboration agreements, our counterparties may have the right to terminate these agreements, in which event we might not be able to develop, manufacture, or market any product that is covered by these agreements or may face other penalties under the agreements. Such an occurrence could diminish the value of the product candidate being developed under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or reinstated agreements with less favorable terms or cause us to lose our rights under these agreements, including our rights to important intellectual property or technology.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property or claiming ownership of what we regard as our own intellectual property.
Some of our employees were previously employed at universities or other biotechnology or pharmaceutical companies. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that these employees or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. Litigation may be necessary to defend against these claims.
34
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our and their assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to management.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our ability to compete in the marketplace.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and product candidates, we also rely on trade secrets, including unpatented know-how, technology, and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors, and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the U.S. are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
35
Risks Related to Regulatory Approval of Our Product Candidates and Other Legal Compliance Matters
If we are not able to obtain required regulatory approvals, we will not be able to commercialize KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop, and our ability to generate revenue will be materially impaired. The marketing approval process is expensive, time-consuming, and uncertain. As a result, we cannot predict when or if we, or any collaborators we may have in the future, will obtain marketing approval to commercialize KIO-101, KIO-201, KIO-301, or any other product candidate.
The activities associated with the development and commercialization of our product candidates, including KIO-101, KIO-201, and KIO-301, including design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale, and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the U.S. and similar regulatory authorities outside the U.S. Failure to obtain marketing approval for a product candidate will prevent us from commercializing the product candidate. We have not received approval to market KIO-101, KIO-201, KIO-301, or any other product candidate from regulatory authorities in any jurisdiction. We have only limited experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on third-party CROs and consultants to assist us in this process.
The process of obtaining marketing approvals, both in the U.S. and abroad, is expensive and may take many years, especially if additional clinical trials are required, if approval is obtained at all. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s safety, purity, and potency. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory authorities. The FDA or other regulatory authorities may determine that KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop is not safe, effective or pure, is only moderately effective or has undesirable or unintended side effects, toxicities, or other characteristics that preclude our obtaining marketing approval or prevent or limit commercial use. Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
The regulatory process can vary substantially based upon a variety of factors, including the type, complexity, and novelty of the product candidates involved.
Failure to obtain marketing approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In order to market and sell KIO-101, KIO-201, KIO-301, and any other product candidate that we may develop in other jurisdictions, we or our third-party collaborators must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the U.S. generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the U.S., it is required that the product be approved for reimbursement before the product can be sold in that country. We or these third parties may not obtain approvals from regulatory authorities outside the U.S. on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the U.S. does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market.
36
Even if we, or any collaborators we may have in the future, obtain marketing approvals for KIO-101, KIO-201, KIO-301, or our other product candidates, the terms of those approvals, ongoing regulations and post-marketing restrictions may limit how we, or they, manufacture and market our products, which could materially impair our ability to generate revenue.
Once marketing approval has been granted, an approved product and its manufacturer and marketer are subject to ongoing review and extensive regulation. We, and any collaborators we may have in the future, must therefore comply with requirements concerning advertising and promotion for any of our products for which we or they obtain marketing approval. Promotional communications with respect to prescription products are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved labeling. Thus, if KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop receives marketing approval, the accompanying label may limit the approved use of our product, which could limit sales of the product.
In addition, manufacturers of approved products and those manufacturers’ facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to CGMPs, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation and reporting requirements. We, our contract manufacturers, our future collaborators, and their contract manufacturers will also be subject to other regulatory requirements, including submissions of safety and other post-marketing information and reports, registration and listing requirements, requirements regarding the distribution of samples to physicians, recordkeeping, and costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of the product, such as the requirement to implement a risk evaluation and mitigation strategy.
We may be subject to substantial penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our products.
Violations of the Federal Food, Drug, and Cosmetic Act relating to the promotion or manufacturing of prescription products may lead to investigations by the FDA, Department of Justice, and state Attorneys General alleging violations of federal and state healthcare fraud and abuse laws, as well as state consumer protection laws. In addition, later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may yield various adverse results, including:
•restrictions on such products, manufacturers, or manufacturing processes;
•restrictions on the labeling or marketing of a product;
•restrictions on product distribution or use;
•requirements to conduct post-marketing studies or clinical trials;
•warning letters;
•withdrawal of the products from the market;
•refusal to approve pending applications or supplements to approved applications that we submit;
•recall of products;
•fines, restitution, or disgorgement of profits or revenues;
•suspension or withdrawal of marketing approvals;
•refusal to permit the import or export of our products;
•product seizure; or
•injunctions or the imposition of civil or criminal penalties.
37
Our relationships with customers and third-party payors may be subject, directly or indirectly, to applicable anti-kickback, fraud and abuse, false claims, transparency, health information privacy and security, and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, administrative burdens, and diminished profits and future earnings.
Healthcare providers, physicians, and third-party payors in the U.S. and elsewhere will play a primary role in the recommendation and prescription of any product candidates, including KIO-101, KIO-201, and KIO-301, for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell, and distribute any products for which we obtain marketing approval. In addition, we may be subject to transparency laws and patient privacy regulation by U.S. federal and state governments and by governments in foreign jurisdictions in which we conduct our business. The applicable federal, state, and foreign healthcare laws and regulations that may affect our ability to operate include:
•the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid;
•federal civil and criminal false claims laws and civil monetary penalty laws, including the federal False Claims Act, which impose criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
•the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and their respective implementing regulations, which imposes obligations, including mandatory contractual terms, on covered healthcare providers, health plans and healthcare clearinghouses, as well as their business associates, with respect to safeguarding the privacy, security, and transmission of individually identifiable health information; and
•analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state and foreign laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations may involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations, or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal, and administrative penalties, including, without limitation, damages, fines, imprisonment, exclusion from participation
38
in government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, it may be subject to criminal, civil, or administrative sanctions, including exclusions from participation in government funded healthcare programs.
Previously enacted and future legislation may affect our ability to commercialize and the prices we obtain for any products that are approved in the U.S. or foreign jurisdictions.
In the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could affect our ability to profitably sell or commercialize any product candidate, including KIO-101, KIO-201, and KIO-301, for which we obtain marketing approval or that we may in-license. The pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by legislative initiatives. Current laws, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product.
In the U.S., the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA) changed the way Medicare covers and pays for pharmaceutical products. Cost reduction initiatives and other provisions of this legislation could limit coverage of and reduce the price that we receive for any approved products. While the MMA applies only to product benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from the MMA or other healthcare reform measures may result in a similar reduction in payments from private payors.
In March 2010, former President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively “PPACA”). Among the provisions of PPACA of importance to our business, including, without limitation, our ability to commercialize and the prices we may obtain for any of our product candidates and that are approved for sale, are the following:
•an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents;
•an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program;
•a new Medicare Part D coverage gap discount program, in which participating manufacturers must agree to offer 50% point-of-sale discounts off negotiated drug prices during the coverage gap period as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D;
•expansion of healthcare fraud and abuse laws, including the federal False Claims Act and the federal Anti-Kickback Statute, and the addition of new government investigative powers, and enhanced penalties for noncompliance;
•extension of manufacturers’ Medicaid rebate liability;
•expansion of eligibility criteria for Medicaid programs; and
•expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program.
In addition, other legislative changes have been proposed and adopted since PPACA was enacted. These changes include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect in April 2013, and will remain in effect through 2030 unless additional Congressional action is taken. In January 2013, former President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in
39
additional reductions in Medicare and other healthcare funding. In addition, it is possible that changes in administration and policy could result in additional proposals and/or changes to health care system legislation.
Additionally, in light of the rising cost of prescription drugs and biologics, there has been heightened governmental scrutiny in the U.S. of pharmaceutical pricing practices. Such scrutiny has resulted in several recent congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. At both the federal and state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures. In some cases, the legislation and regulations are designed to encourage importation from other countries and bulk purchasing.
We expect that these, and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved drug. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our drugs.
The pricing of prescription pharmaceuticals is also subject to governmental control outside the U.S. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our ability to generate revenues and become profitable could be impaired.
If we or our third-party manufacturers fail to comply with environmental, health, and safety laws and regulations, we could become subject to fines or penalties or incur significant costs.
We and our third-party manufacturers are subject to numerous environmental, health, and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment, and disposal of hazardous materials and wastes. From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees, this insurance may not provide adequate coverage against potential liabilities.
In addition, we may incur substantial costs in order to comply with current or future environmental, health, and safety laws and regulations. These current or future laws and regulations may impair our research, development, or production efforts. Our failure to comply with these laws and regulations also may result in substantial fines, penalties, or other sanctions.
Further, with respect to the operations of our third-party contract manufacturers, it is possible that if they fail to operate in compliance with applicable environmental, health, and safety laws and regulations or properly dispose of wastes associated with our products, we could be held liable for any resulting damages, suffer reputational harm, or experience a disruption in the manufacture and supply of our product candidates or products.
40
Risks Related to Employee Matters and Managing Growth
Our future success depends on our ability to retain key executives and to attract, retain, and motivate qualified personnel.
We are highly dependent on the research and development, clinical, and business development expertise of Brian M. Strem, our Chief Executive Officer, as well as the other principal members of our management, scientific, and clinical team and a number of third-party consultants. Although we have entered into an employment agreement with Dr. Strem, he may terminate his employment with us at any time. We do not maintain “key person” insurance for any of our executives or other employees.
Recruiting and retaining qualified scientific, clinical, manufacturing, and sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development, and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of, and commercialize products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain, or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. The availability of qualified personnel in the markets in which we operate has declined in recent years and competition for such labor has increased. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
We expect to expand our development capabilities and potentially implement sales, marketing, and distribution capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We may experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of drug development, regulatory affairs and, if any of our product candidates receives marketing approval, sales, marketing, and distribution. To manage our potential future growth, we must continue to implement and improve our managerial, operational, and financial systems, expand our facilities, and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a company with such potential growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We may fail to realize any benefits and incur losses related to any acquisition.
We regularly explore opportunities to grow our business, including through acquiring companies. The success of our strategic acquisitions will depend, in part, on our ability to successfully integrate the acquired businesses with our existing business. It is possible that the integration process could result in the loss of key employees; the disruption of ongoing business; or inconsistencies in standards, controls, procedures, and policies that adversely affect our ability to maintain relationships with vendors, customers, and employees or to achieve the anticipated benefits of the acquisition. Successful integration may also be hampered by any differences between the operations and corporate culture of the two organizations. If we experience difficulties with the integration process, the anticipated benefits of the acquisition may not be realized fully, or at all, or may take longer to realize than expected.
41
Risks Related to Our Common Stock
Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, any future debt agreements that we may enter into, may preclude us from paying dividends without the lenders’ consent or at all. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
We could face delisting from Nasdaq in the event we do not meeting its minimum bid price rules.
On February 23, 2022, we received a written notification (the “Notice Letter”) from Nasdaq indicating that we were not in compliance with Nasdaq Listing Rule 5450(a)(1), as the closing bid price for our common stock was below the $1.00 per share requirement for the last 30 consecutive business days. The Notice Letter stated that we have 180 calendar days, or until August 22, 2022 (the “Initial Compliance Period”), to regain compliance with the minimum bid price requirement. In accordance with Nasdaq Listing Rule 5810(c)(3)(A), we can regain compliance if the closing bid price of our common stock is at least $1.00 for a minimum of 10 consecutive business days.
On August 23, 2022, Nasdaq notified us in writing (the “Extension Letter”) that while we had not regained compliance with the Bid Price Rule, we were eligible for an additional 180-day compliance period, or until February 20, 2023, to regain compliance with the Bid Price Rule. Nasdaq’s determination was based on our having met the continued listing requirement for market value of publicly held shares and all other applicable requirements for initial listing on The Nasdaq Capital Market, with the exception of the Bid Price Rule, and on our written notice to Nasdaq of its intention to cure the deficiency during the second compliance period by effecting a reverse stock split, if necessary.
On October 12, 2022, we received a letter from Nasdaq notifying us that the closing bid price of our common stock had been at $1.00 per share or greater for the last 10 consecutive business days and we had regained compliance with the Bid Price Rule and this matter had been closed.
In the event that we lose compliance with Listing Rule 5450(a)(1) again and do not regain compliance prior to the expiration of the compliance period, we will receive written notification that our securities are subject to delisting. At that time, we may appeal the delisting determination to a hearings panel pursuant to the procedures set forth in the applicable Nasdaq Listing Rules. A delisting of our common stock would have an adverse effect on the market liquidity of our common stock and, as a result, the market price for our common stock could become more volatile. Further, a delisting also could make it more difficult for us to raise additional capital. We intend to monitor the closing bid price of our common stock and may conduct a reverse stock split, if necessary, to regain compliance with the Nasdaq bid price rule.
We have identified material weaknesses in our internal control over financial reporting. If we are unable to remediate these material weaknesses, or if we experience additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls, we may not be able to accurately or timely satisfy the requirements applicable to public companies, which may adversely affect investor confidence in us, and, as a result, the market price of our common stock.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with GAAP. Our management is likewise required, on a quarterly basis, to evaluate the effectiveness of our internal controls and to disclose any changes and material weaknesses identified through such evaluation in those internal controls. A material weakness is a deficiency, or combination of deficiencies in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our consolidated financial statements will not be prevented or detected on a timely basis.
42
We have previously identified the following material weaknesses because we did not maintain effective controls regarding:
•information technology general controls (“ITGC”) which could result in misstatements potentially impacting all financial statement accounts and disclosures. Specifically, user access controls were not appropriately designed and maintained to adequately restrict network user and privileged access to the appropriate personnel. In addition, there was a lack of sufficient segregation of duties.
•the design and maintenance of formal accounting policies and processes to analyze, account for and disclose significant and unusual transactions in accordance with GAAP.
•the execution of controls with a sufficient level of precision over the completeness and accuracy of financial information used in the preparation of our financial statements.
To respond to these material weaknesses, we have devoted, and plan to continue to devote, significant effort and resources to the remediation and improvement of our internal control over financial reporting. Our remediation efforts include the establishment and maintenance of accounting policies, procedures and controls to account for and disclose significant unusual transactions, the engagement of resources for technical advisory services, and the implementation of a new accounting system with greater automation. The elements of our remediation plan can only be accomplished over time, and we can offer no assurance that these initiatives will ultimately have the intended effects.
Any failure to maintain such internal control could adversely impact our ability to report our financial position and results from operations on a timely and accurate basis. If our consolidated financial statements are not accurate, investors may not have a complete understanding of our operations. Likewise, if our consolidated financial statements are not filed on a timely basis, we could be subject to sanctions or investigations by the stock exchange on which our ordinary shares and other securities are listed, the SEC or other regulatory authorities. In either case, there could result a material adverse effect on our business. Ineffective internal controls could also cause investors to lose confidence in our reported financial information which could have a negative effect on the trading price of our stock.
We can give no assurance that the measures we have taken and plan to take in the future will remediate the material weaknesses identified or that any additional material weaknesses or restatements of financial results will not arise in the future due to a failure to implement and maintain adequate internal control over financial reporting or circumvention of these controls. In addition, even if we are successful in strengthening our controls and procedures, in the future those controls, and procedures may not be adequate to prevent or identify irregularities or errors or to facilitate the fair presentation of our consolidated financial statements.
General Risk Factors
The coronavirus pandemic could adversely impact our business, including clinical trials.
In December 2019, a novel strain of coronavirus, COVID-19, was reported to have surfaced in Wuhan, China. Since then, COVID-19 has spread globally. As the COVID-19 pandemic continues, we could experience disruptions that could impact our business and clinical trials, including:
•delays or difficulties in enrolling patients in clinical trials;
•delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
•interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers, and others;
•limitations in employee resources that would otherwise be focused on the conduct of clinical trials, including sickness of employees or their families or the desire of employees to avoid contact with groups of people;
43
•interruption in global manufacturing and shipping that may affect the transport of clinical trial materials and materials, including testing equipment and personal protective equipment, used at our facilities;
The global outbreak of the COVID-19 coronavirus continues to evolve. The extent to which COVID-19 may impact our business and clinical trials will depend on future developments which are highly uncertain and cannot be predicted with confidence, such as the duration of the pandemic, the emergence of new variants, travel restrictions and social distancing in the U.S. and other countries, business closures or business disruptions, and the effectiveness of actions taken in the U.S. and other countries to contain and treat the disease.
Laws and regulations governing international operations may preclude us from developing, manufacturing, and selling certain products outside of the U.S. and require us to develop and implement costly compliance programs.
We must dedicate additional resources to comply with numerous laws and regulations in each jurisdiction in which we plan to operate, including our operations in Australia and Austria. The Foreign Corrupt Practices Act (FCPA) prohibits any U.S. individual or business from paying, offering, authorizing payment, or offering of anything of value, directly or indirectly, to any foreign official, political party, or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the U.S. to comply with certain accounting provisions requiring the Company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Various laws, regulations, and executive orders also restrict the use and dissemination outside of the U.S., or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. Our foreign operations require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and product candidates outside of the U.S., which could limit our growth potential and increase our development costs.
The failure to comply with laws governing international business practices may result in substantial civil and criminal penalties and suspension or debarment from government contracting. The SEC also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA’s accounting provisions.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems, and those of our CROs and other third parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed.
44
Provisions in our corporate charter documents and under Delaware law could make an acquisition of our company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our restated certificate of incorporation and our amended and restated bylaws may discourage, delay, or prevent a merger, acquisition, or other change in control of our company that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions:
•establish a classified board of directors such that only one of three classes of directors is elected each year;
•allow the authorized number of our directors to be changed only by resolution of our board of directors;
•limit the manner in which stockholders can remove directors from our board of directors;
•establish advance notice requirements for stockholder proposals that can be acted on at stockholder meetings and nominations to our board of directors;
•require that stockholder actions must be effected at a duly called stockholder meeting and prohibit actions by our stockholders by written consent;
•limit who may call stockholder meetings;
•authorize our board of directors to issue preferred stock without stockholder approval, which could be used to institute a “poison pill” that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our board of directors; and
•require the affirmative vote of stockholders holding at least two-thirds of shares entitled to be cast to amend or repeal specified provisions of our restated certificate of incorporation or our amended and restated bylaws.
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock.
Our stock price may be volatile. The stock market in general and the market for smaller specialty pharmaceutical companies in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, you may not be able to sell your common stock at or above the price you paid for such shares. The market price for our common stock may be influenced by many factors, including:
•the success of competitive products or technologies;
•results of clinical trials of KIO-101, KIO-201, KIO-301, or any other product candidate that we may develop;
•results of clinical trials of product candidates of our competitors;
•regulatory or legal developments in the U.S. and other countries;
45
•developments or disputes concerning patent applications, issued patents or other proprietary rights;
•the recruitment or departure of key scientific or management personnel;
•the level of expenses related to any of our product candidates or clinical development programs;
•the results of our efforts to discover, develop, acquire, or in-license additional products, product candidates, or technologies for the treatment of ophthalmic diseases, the costs of commercializing any such products, and the costs of development of any such product candidates or technologies;
•actual or anticipated changes in estimates as to financial results, development timelines, or recommendations by securities analysts;
•variations in our financial results or those of companies that are perceived to be similar to us;
•changes in the structure of healthcare payment systems;
•reduction in stock price could indicate impairment of the goodwill and intangible assets;
•market conditions in the pharmaceutical and biotechnology sectors;
•general economic, industry, and market conditions; and
•the other factors described in this “Risk Factors” section.
In the past, following periods of volatility in the market price of a company’s securities, securities class-action litigation has often been instituted against that company. We also may face securities class-action litigation if we cannot obtain regulatory approvals for or if we otherwise fail to commercialize KIO-101, KIO-201, or KIO-301. Such litigation, if instituted against us, could cause us to incur substantial costs to defend such claims and divert management’s attention and resources.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As of December 31, 2022, we had federal net operating loss carryforwards of approximately $80.5 million, state net operating loss carryforwards of approximately $52.6 million, and aggregate federal and state research and development tax credit carryforwards of approximately $2.5 million and $0.5 million, respectively, available to reduce future taxable income. Certain of these federal and state net operating loss carryforwards and federal and state tax credit carryforwards will expire at various dates through 2041, if not utilized. Federal net operating losses generated as of December 31, 2017, will carry-forward until 2037 and net operating losses generated during the year ended December 31, 2018, and later will be carried forward indefinitely until utilized, but their utilization will be limited to 80% of taxable income. Utilization of these net operating loss and tax credit carryforwards may be subject to a substantial limitation under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, and comparable provisions of state, local, and foreign tax laws due to changes in ownership of our company that have occurred previously or that could occur in the future. Under Section 382 of the Code and comparable provisions of state, local, and foreign tax laws, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change by value in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes, such as research and development tax credits, to reduce its post-change income may be limited. We have not completed a study to determine whether our initial public offering, subsequent public and private offerings, and other transactions that have occurred may have triggered an ownership change limitation. We may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership. As a result, if we generate taxable income, our ability to use our pre-change net operating loss and tax credits carryforwards to reduce U.S. federal and state taxable income may be subject to limitations, which could result in increased future tax liability to us. In addition, the Tax Cuts and Jobs Act (TCJA) enacted on December 22, 2017, limits the amount of net operating losses that we are permitted to deduct in any taxable year to 80% of our taxable income in such year. The TCJA also eliminates the ability to carry back net operating losses to prior years, but allows net operating losses generated after 2017 to be carried forward indefinitely. As such, there is a risk that due to such items, our existing net operating losses could
46
expire or be unavailable to offset future income. Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Sales of a substantial number of shares of our common stock by our existing stockholders in the public market could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur, could significantly reduce the market price of our common stock and impair our ability to raise adequate capital through the sale of additional equity securities.
We are a smaller reporting company and the reduced disclosure requirements applicable to smaller reporting companies may make our common stock less attractive to investors.
We are a smaller reporting company (SRC) and a non-accelerated filer, which allows us to take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not SRCs or non-accelerated filers, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended, reduced disclosure obligations, including disclosures regarding executive compensation, in our Annual Report and our periodic reports and proxy statements and providing only two years of audited consolidated financial statements in our Annual Report and our periodic reports. We will remain an SRC until (a) the aggregate market value of our outstanding common stock held by non-affiliates as of the last business day our most recently completed second fiscal quarter exceeds $250 million or (b) in the event we have over $100 million in annual revenues, the aggregate market value of our outstanding common stock held by non-affiliates as of the last business day our most recently completed second fiscal quarter exceeds $700 million. We cannot predict whether investors will find our common stock less attractive if we rely on certain or all of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile and may decline.
We incur increased costs as a result of operating as a public company, and our management is required to devote substantial time to new compliance initiatives and corporate governance practices.
As a public company, we incur significant legal, accounting, and other expenses that we did not incur as a private company. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, Financial Industry Regulatory Authority (FINRA) rules, and other applicable securities rules and regulations impose various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase our legal and financial compliance costs and make some activities more time-consuming and costly.
We continue to evaluate these rules and regulations and cannot predict or estimate the amount of additional costs we may incur or the timing of such costs. These rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Pursuant to Section 404 of the Sarbanes-Oxley Act, or Section 404, we are required to furnish a report by our management on our internal control over financial reporting. However, while we remain a non-accelerated filer, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we have engaged in a process to document and evaluate our internal control over financial reporting. In this regard, we will need to continue to dedicate internal resources, engage outside consultants, and adopt a detailed work plan to continue to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a continuous reporting and improvement process for internal control over financial reporting. If we identify one or more material weaknesses in our internal control over financial reporting, it could
47
result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our consolidated financial statements.
A material amount of our assets represents intangible assets, and our net income would be reduced if our intangible assets become impaired.
As of December 31, 2022, intangible assets, net, represented approximately $10.7 million, or 58% of our total assets. Goodwill is generated in our acquisitions when the cost of an acquisition exceeds the fair value of the net tangible and identifiable intangible assets we acquire. Goodwill and indefinite-lived intangible assets are subject to an impairment analysis at least annually based on fair value. Intangible assets relate primarily to in-process research and development (IPR&D) and patents acquired by us as part of our acquisitions of other companies, and are subject to an impairment analysis whenever events or changes in circumstances exist that indicate that the carrying value of the intangible asset might not be recoverable. If market and economic conditions or business performance deteriorate, the likelihood that we would record an impairment charge would increase, which impairment charge could materially and adversely affect our financial condition and operating results.
Risks Relating to This Offering
We have broad discretion to determine how to use the proceeds raised in this offering, and we may not use the proceeds effectively.
Our management will have broad discretion over the use of proceeds from this offering, and we could spend the proceeds from this offering in ways with which you may not agree or that do not yield a favorable return. We intend to use the net proceeds from this offering to support our operations, including for clinical trials, for working capital and for other general corporate purposes. If we do not invest or apply the proceeds of this offering in ways that improve our operating results, we may fail to achieve expected financial results, which could cause our stock price to decline.
You will experience immediate and substantial dilution when you purchase shares in this offering.
You will incur immediate and substantial dilution as a result of this offering. After giving effect to the assumed sale by us of 2,582,160 shares of our common stock in this offering at the assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant, the last reported sale price of our common stock on The Nasdaq Capital Market on May 30, 2023, assuming conversion of all shares of Series F Preferred Stock into shares of common stock, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, investors in this offering will suffer an immediate dilution of $1.18 per share. The final public offering price will be determined through negotiations between us and the underwriters in the offering and may be at a discount to the current market price. Therefore, the assumed public offering price used throughout this prospectus may not be indicative of the final public offering price. Furthermore, if the underwriters exercise their option to purchase additional shares of common stock and/or warrants to purchase shares of common stock, you will experience further dilution.
If we issue additional common stock, or securities convertible into or exchangeable or exercisable for common stock, our stockholders, including investors who purchase shares of common stock in this offering, may experience additional dilution, and any such issuances may result in downward pressure on the price of our common stock. We may not be able to sell shares or other securities in any other offering at a price per share that is equal to or greater than the price per share paid by investors in this offering, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders. See the section entitled “Dilution” below for a more detailed discussion of the dilution you will incur if you purchase common stock in this offering.
48
You will experience immediate and substantial dilution in the net tangible book value per share of the Series F Preferred Stock you purchase.
Since the price per share of our Series F Preferred Stock being offered is substantially higher than the net tangible book per share of our underlying common stock, you will suffer substantial dilution in the net tangible book value of the shares that you purchase in this offering. Based on the assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant, the last reported sale price of our common stock on The Nasdaq Capital Market on May 30, 2023, if you purchase Series F Preferred Stock in this offering, you will suffer immediate and substantial dilution of $1.18 per share in the net tangible book value of the shares of common stock underlying the Series F Preferred Stock. See the section entitled “Dilution” below for a more detailed discussion of the dilution you will incur if you purchase Series F Preferred Stock in this offering.
The issuance of additional equity securities may negatively impact the trading price of our common stock.
We have issued equity securities in the past, will issue equity securities in this offering and expect to continue to issue equity securities to finance our activities in the future. In addition, outstanding options and warrants to purchase our common stock may be exercised and additional options and warrants may be issued, resulting in the issuance of additional shares of common stock. The issuance by us of additional equity securities, including the shares of common stock issuable upon exercise of the warrants issued by us in this offering, would result in dilution to our stockholders, and even the perception that such an issuance may occur could have a negative impact on the trading price of our common stock.
There is no public market for the Series F Preferred Stock, the Class C Warrants or the Class D Warrants being offered by us in this offering.
There is no established public trading market for the Series F Preferred Stock, the Class C Warrants or the Class D Warrants being offered in this offering, and we do not expect a market to develop. In addition, we do not intend to apply to list the Series F Preferred Stock, the Class C Warrants or the Class D Warrants on any national securities exchange or other nationally recognized trading system, including The Nasdaq Capital Market. Without an active market, the liquidity of the Series F preferred stock, the Class C Warrants and the Class D Warrants will be limited.
The Class C Warrants and Class D Warrants are speculative in nature.
The Class C Warrants and the Class D Warrants do not confer any rights of common stock ownership on its holders, such as voting rights or the right to receive dividends, but rather merely represent the right to acquire shares of common stock at a fixed price for a limited period of time. Specifically, commencing on the date of issuance, holders of the Class C Warrants may exercise their right to acquire the common stock and pay an exercise price of $ per share, subject to certain adjustments, prior to the five year anniversary from the date of issuance with respect to the Class C Warrants and the one year anniversary from the date of issuance with respect to the Class D Warrants, after which date any unexercised warrants will expire and have no further value. Moreover, following this offering, the market value of the Class C Warrants and the Class D Warrants, if any, is uncertain and there can be no assurance that the market value of the Class C Warrants and the Class D Warrants will equal or exceed their imputed offering price. The Class C Warrants and the Class D Warrants will not be listed or quoted for trading on any market or exchange. There can be no assurance that the market price of the common stock will ever equal or exceed the exercise price of the Class C Warrants and the Class D Warrants, and consequently, whether it will ever be profitable for holders of the Class C Warrants and the Class D Warrants to exercise the warrants.
A substantial number of shares of our common stock may be sold in this offering, which could cause the price of our common stock to decline.
In this offering, we will sell shares of common stock and shares of Series F Preferred Stock convertible into up to shares of common stock, collectively representing approximately % of our outstanding common stock as of , 2023. This sale and any future sales of a substantial number of shares of our common stock in the public market, or the perception that such sales may occur, could adversely affect the price of our common stock. We cannot predict the effect, if any, that market sales of those shares of common stock or the availability of those shares of common stock for sale will have on the market price of our common stock.
49
A significant number of additional shares of our common stock may be issued upon the conversion of existing securities, including the Series F Preferred Stock, which issuances would substantially dilute existing stockholders and may depress the market price of our common stock.
As of May 30, 2023, there were 2,024,270 shares of common stock outstanding, shares of Series D Convertible Preferred Stock that are convertible into 52 shares of common stock, 1,613,483 shares of common stock underlying outstanding warrants, 202,126 shares of common stock underlying outstanding options, and 67,250 shares of common stock granted through a restricted stock award. In addition, shares of common stock will be issuable upon conversion of our Series F Preferred Stock. The issuance of any such shares of common stock would substantially dilute the proportionate ownership and voting power of existing security holders, and their issuance, or the possibility of their issuance, may depress the market price of our common stock.
Upon conversion of the Series F Preferred Stock, holders may receive less valuable consideration than expected because the value of our common stock may decline after such holders exercise their conversion right but before we settle our conversion obligation.
Under the Series F Preferred Stock, a converting holder will be exposed to fluctuations in the value of our common stock during the period from the date such holder surrenders shares of Series F Preferred Stock for conversion until the date we settle our conversion obligation. Upon conversion, we will be required to deliver the shares of our common stock, together with a cash payment for any fractional share (if so elected by the Company), on the second business day following the relevant conversion date. Accordingly, if the price of our common stock decreases during this period, the value of the shares of common stock that you receive will be adversely affected and would be less than the conversion value of the Series F Preferred Stock on the conversion date.
We may issue additional series of preferred stock that rank senior or equally to the Series F Preferred Stock as to dividend payments and liquidation preference.
Neither our restated certificate of incorporation nor the Certificate of Designation for the Series F Preferred Stock prohibits us from issuing additional series of preferred stock that would rank senior or equally to the Series F Preferred Stock as to dividend payments and liquidation preference. Our restated certificate of incorporation provides that we have the authority to issue up to 10,000,000 shares of preferred stock. The issuances of other series of preferred stock could have the effect of reducing the amounts available to the Series F Preferred Stock in the event of our liquidation, winding-up or dissolution. It may also reduce cash dividend payments on the Series F Preferred Stock if we do not have sufficient funds to pay dividends on all Series F Preferred Stock outstanding and outstanding parity preferred stock.
Future issuances of preferred stock may adversely affect the market price for our common stock.
Additional issuances and sales of preferred stock, or the perception that such issuances and sales could occur, may cause prevailing market prices for our common stock to decline and may adversely affect our ability to raise additional capital in the financial markets at times and prices favorable to us.
50
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains, and the documents incorporated herein by reference contain, forward-looking statements that involve risks and uncertainties. The forward-looking statements are contained principally in the sections of this prospectus and the documents incorporated herein by reference under the captions “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business.” In some cases, you can identify forward-looking statements by terms such as “may,” “might,” “will,” “objective,” “intend,” “should,” “seek,” “aim,” “think,” “optimistic,” “strategy,” “goals,” “sees,” “new,” “guidance,” “future,” “continue,” “drive,” “growth,” “long-term,” “develop,” “possible,” “emerging,” “opportunity,” “pursue,” “could,” “can,” “would,” “expect,” “believe,” “anticipate,” “project,” “target,” “design,” “estimate,” “predict,” “potential,” “plan” or the negative of these terms, and similar expressions intended to identify forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
•the timing and success of preclinical studies and clinical trials conducted by us and our development partners;
•the ability to obtain and maintain regulatory approval of our product candidates, and the labeling for any approved products;
•the scope, progress, expansion, and costs of developing and commercializing our product candidates;
•the size and growth of the potential markets for our product candidates and the ability to serve those markets;
•our expectations regarding our expenses and revenue, the sufficiency of our cash resources and needs for additional financing;
•the rate and degree of market acceptance of any of our product candidates;
•our expectations regarding competition;
•our anticipated growth strategies;
•our ability to attract or retain key personnel;
•our ability to establish and maintain development partnerships;
•our expectations regarding federal, state and foreign regulatory requirements;
•regulatory developments in the U.S. and foreign countries;
•our ability to obtain and maintain intellectual property protection for our product candidates;
•the anticipated trends and challenges in our business and the market in which we operate;
•the impact of the evolving COVID-19 pandemic and the global response thereto; and
•our use of proceeds from this offering.
Any forward-looking statement made by us in this prospectus speaks only as of the date on which it is made. Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties. Except as required by
51
applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
52
MARKET FOR COMMON STOCK
Our common stock is listed on The Nasdaq Capital Market under the symbol “KPRX”. On May 30, 2023, the last reported sale price of our common stock as reported by The Nasdaq Capital Market was $2.13 per share. As of such date, we had approximately 47 stockholders of record.
53
DIVIDEND POLICY
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain future earnings, if any, and all currently available funds for use in the operation of our business and do not anticipate paying any cash dividends in the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our Board of Directors after considering our financial condition, results of operations, capital requirements, business prospects and other factors the Board of Directors deems relevant, and subject to the restrictions contained in our current or future financing instruments.
54
USE OF PROCEEDS
We estimate the net proceeds from this offering will be approximately $4.8 million, after deducting estimated underwriting discounts and commissions and our estimated offering expenses, and based on the assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant (the last reported sale price of our common stock on The Nasdaq Capital Market on May 30, 2023) assuming no exercise of the underwriters’ over-allotment option.
A $0.25 increase (decrease) in the assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant would increase (decrease) the net proceeds to us from this offering by approximately $0.6 million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
Similarly, a 1,000,000 share increase in the number of shares of common stock, an increase of 1,000,000 in the number of Class C Warrants offered by us, and an increase of 1,000,000 in the number of Class D Warrants offered by us, as set forth on the cover page of this prospectus, would increase the net proceeds to us by $1.7 million, assuming the assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant remains the same, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Likewise, a 1,000,000 share decrease in the number of shares of common stock, a decrease of 1,000,000 in the number of Class C Warrants offered by us, and a decrease of 1,000,000 in the number of Class D Warrants offered by us, as set forth on the cover page of this prospectus, would decrease the net proceeds to us by $2.0 million, assuming the assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant remains the same, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
We intend to use the net proceeds from this offering, together with other available funds, to support our operations, including for clinical trials, for working capital and for other general corporate purposes. We have not yet determined the amount of net proceeds to be used specifically for any of the foregoing purposes.
Pending use of the proceeds as described above, we intend to invest the net proceeds of this offering in short-term, interest-bearing, investment-grade securities or certificates of deposit.
The amounts and timing of our actual expenditures will depend on numerous factors, including the progress of our clinical trials, as well as the amount of cash used in our operations. We may find it necessary or advisable to use the net proceeds for other purposes, and we will have broad discretion in the application of the net proceeds and investors will be relying on the judgment of our management regarding the application of the net proceeds from this offering.
Based upon our historical and anticipated future growth and our financial needs, we may engage in additional financings of a character and amount that we determine as the need arises. We may raise additional capital through additional public or private financings, the incurrence of debt and other available sources.
55
DILUTION
If you purchase our common stock, Series F Preferred Stock, or both, in this offering, assuming the conversion of the Series F Preferred Stock into shares of our common stock, you will experience dilution to the extent of the difference between the public offering price per share in this offering and our as adjusted net tangible book value per share immediately after this offering. Net tangible book value (deficit) per share is equal to the amount of our total tangible assets, less total liabilities, divided by the number of outstanding shares of our common stock. As of March 31, 2023, our net tangible book value was $(581,405), or approximately $(0.30) per share.
After giving effect to the sale of 2,582,160 shares of common stock by us at an assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant (the last reported sale price of our common stock on The Nasdaq Capital Market on May 30, 2023), assuming that all shares of Series F Preferred Stock are converted into shares of common stock excluding shares that may be issued upon exercise of the underwriters’ over-allotment option and after deducting estimated underwriting discounts and commissions and estimated offering expenses, our as adjusted net tangible book value as of March 31, 2023 would have been approximately $4.3 million, or $0.95 per share of common stock, which excludes the Class C Warrants to purchase 2,582,160 shares of our common stock and the Class D Warrants to purchase 2,582,160 shares of our common stock to be issued to investors in this offering. This represents an immediate increase in net tangible book value of $1.25 per share of common stock to existing stockholders and immediate dilution of $1.18 per share of common stock to investors purchasing our common stock in this offering at the assumed public offering price. The final combined public offering price will be determined between us and the underwriters in the offering and may be at a discount to the current market price. Therefore, the assumed combined public offering price used throughout this prospectus may not be indicative of the final combined public offering price. The following table illustrates this dilution on a per share basis:
| Assumed combined public offering price per share of common stock and warrants | $ | 2.13 | |||
| Net tangible book value per share as of March 31, 2023 | $ | (0.30) | |||
| Increase in net tangible book value per share after giving effect to this offering | $ | 1.25 | |||
| As adjusted net tangible book value per share after giving effect to this offering | $ | 0.95 | |||
| Dilution per share to new investors | $ | 1.18 | |||
The information above is illustrative only and will change based on actual pricing and other terms of this offering determined at pricing. Each $0.25 increase (decrease) in the assumed combined public offering price of $2.13 per share of common stock, Class C Warrant and Class D Warrant would increase (decrease) the as adjusted net tangible book value by $0.13 per share of common stock and the dilution to new investors by $0.12 per share, assuming that the number of shares and warrants offered by us, as set forth on the cover page of this prospectus, remains the same, after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
We may also increase or decrease the number of shares we are offering. An increase of 1,000,000 shares in the number of shares of common stock, an increase of 1,000,000 in the number of Class C Warrants offered by us, and an increase of 1,000,000 in the number of Class D Warrants offered by us, as set forth on the cover page of this prospectus, would increase our as adjusted net tangible book value by approximately $1.7 million, or approximately $0.15 per share of common stock, and decrease the dilution per share to investors in this offering by approximately $0.15 per share, assuming that the assumed combined public offering price per share of common stock, Class C Warrant and Class D Warrant remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, a decrease of 1,000,000 shares in the number of shares of common stock, a decrease of 1,000,000 in the number of Class C Warrants offered by us, and a decrease of 1,000,000 in the number of Class D Warrants offered by us, as set forth on the cover page of this prospectus, would decrease our as adjusted net tangible book value by approximately $2.0 million, or approximately $0.30 per share, and increase the dilution per share to investors in this offering by approximately $0.30 per share, assuming that the assumed combined public offering price per share of common stock, Class C Warrant and Class D Warrant remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The information discussed above is illustrative only and will adjust based on the actual
56
public offering price, the actual number of shares of common stock, Series F Preferred Stock, Series C Warrants and Series D Warrants that we offer in this offering, and other terms of this offering determined at pricing.
The above discussion and table do not take into account further dilution to investors purchasing our common stock in this offering that could occur upon the exercise of outstanding options and warrants having a per share exercise price less than the public offering price per share in this offering. To the extent that outstanding options or warrants outstanding as of March 31, 2023 are exercised or other shares are issued, investors purchasing our common stock in this offering will experience further dilution. In addition, we may choose to raise additional capital due to market conditions or strategic considerations even if we believe that we have sufficient funds for our current or future operating plans. To the extent that additional capital is raised through the sale of our common stock, including through the sale of securities convertible into or exchangeable or exercisable for common stock, the issuance of these securities could result in further dilution to our stockholders, including investors purchasing our common stock in this offering.
The table and discussion above are based on 1,919,270 shares of our common stock outstanding as of March 31, 2023, assumes no exercise of the underwriters’ over-allotment option, and assumes that the shares of Series F Preferred Stock sold in the offering have been converted, but does not include, as of such date:
•211,578 shares of common stock issuable upon exercise of options outstanding under our 2005 Equity Incentive Plan and 2014 Equity Incentive Plan, at a weighted-average exercise price of approximately $17.01 per share;
•70,550 shares of common stock granted through a restricted stock award subject to release under our 2005 and 2014 Equity Incentive Plans, at a weighted-average exercise price of approximately $0 per share.
•1,653,202 shares of our common stock issuable upon the exercise of outstanding warrants to purchase shares of our common stock with a weighted-average exercise price of $20.55 per share;
•11,175 shares of common stock reserved for future issuance under our 2014 Equity Incentive Plan;
•191 shares of common stock reserved for future issuance under our 2014 Employee Stock Purchase Plan;
•52 shares of common stock issuable upon the conversion of outstanding shares of Series D Convertible Preferred Stock;
•2,582,160 shares of common stock issuable upon the exercise of Class C Warrants to be issued to investors in this offering at an exercise price of $ per share; and
•2,582,160 shares of common stock issuable upon the exercise of Class D Warrants to be issued to investors in this offering at an initial exercise price of $ per share.
57
DESCRIPTION OF THE SECURITIES WE ARE OFFERING
General
Our authorized capital stock consists of 50,000,000 shares of common stock, par value $0.01 per share, and 10,000,000 shares of preferred stock, par value $0.01 per share, of which 3,750 are designated as Series A Convertible Preferred Stock, 10,000 are designated as Series B Convertible Preferred Stock, 10,000 are designated as Series C Convertible Preferred Stock, 20,000 are designated as Series D Convertible Preferred Stock, 1,280 are designated as Series E Convertible Preferred Stock, and are designated as Series F Convertible Preferred Stock, which we refer to as our Series F Preferred Stock. The following description summarizes some of the terms of our restated certificate of incorporation and second amended and restated bylaws, but does not purport to be complete and is qualified in its entirety by the provisions of our restated certificate of incorporation and second amended and restated bylaws, copies of which have been filed as exhibits to the registration statement of which this prospectus is a part.
There were 2,024,270 shares of our common stock, no shares of our Series A Convertible Preferred Stock, Series B Convertible Preferred Stock, Series C Convertible Preferred Stock, Series E Convertible Preferred Stock, or Series F Preferred Stock, and 7 shares of our Series D Convertible Preferred Stock (convertible into an aggregate of 52 shares of common stock) outstanding as of May 30, 2023, assuming no exercise of outstanding options or warrants. There were approximately 47 holders of record of our common stock as of May 30, 2023. This number does not include beneficial owners whose shares are held in street name.
As of May 30, 2023, there were 202,126 shares of common stock subject to outstanding options, 67,250 shares of common stock granted through a restricted stock award subject to release and 1,613,483 shares of common stock subject to outstanding warrants.
Common Stock
Voting Rights. Each holder of common stock is entitled to one vote for each share of common stock held on all matters submitted to a vote of the stockholders, including the election of directors. Except as otherwise provided by law or our restated certificate of incorporation or bylaws, all matters other than the election of directors submitted to the stockholders at any meeting shall be decided by the affirmative vote of a majority of the outstanding shares of common stock present in person or represented by proxy at the meeting and entitled to vote thereon. Directors are elected by a plurality of the votes cast at the meeting. Our restated certificate of incorporation and second amended and restated bylaws do not provide for cumulative voting rights. Because of this, the holders of a majority of the shares of common stock entitled to vote in any election of directors can elect all of the directors standing for election, if they should so choose.
Dividends. Subject to preferences that may be applicable to any then outstanding preferred stock, the holders of our outstanding shares of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds. At present, we have no plans to issue dividends.
Liquidation. In the event of our liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities, subject to the satisfaction of any liquidation preference granted to the holders of any outstanding shares of preferred stock.
Other Rights and Preferences. Holders of our common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of our preferred stock that we may designate and issue in the future.
Fully Paid and Nonassessable. All of our outstanding shares of common stock are fully paid and nonassessable.
58
Forum Selection. Our restated certificate of incorporation provides that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim for breach of a fiduciary duty owed by any of our directors, officers or employees to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law, our restated certificate of incorporation or our bylaws or (iv) any action asserting a claim governed by the internal affairs doctrine. This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act, Securities Act, or, in each case, the rules and regulations thereunder, or any other claim for which the U.S. federal courts have exclusive jurisdiction.
Provisions in our restated certificate of incorporation provide that our board of directors is authorized to issue preferred stock in one or more series, to establish the number of shares to be included in each such series and to fix the designation, powers, preferences and rights of such shares and any qualifications, limitations or restrictions thereof. The issuance of preferred stock may have the effect of delaying, deferring or preventing a change in control of our company without further action by the stockholders and may adversely affect the voting and other rights of the holders of common stock. The issuance of preferred stock with voting and conversion rights may adversely affect the voting power of the holders of common stock, including the loss of voting control to others. At present, we have no plans to issue any additional preferred stock.
Series F Convertible Preferred Stock
Rank. The Series F Preferred Stock ranks (1) on parity with our common stock on an “as converted” basis, (2) on parity with our Series A Convertible Preferred Stock, Series B Convertible Preferred Stock, Series C Convertible Preferred Stock, Series D Convertible Preferred Stock, and Series E Convertible Preferred Stock, (3) senior to any series of our capital stock hereafter created specifically ranking by its terms junior to the Series F Preferred Stock, (4) on parity with any series of our capital stock hereafter created specifically ranking by its terms on parity with the Series F Preferred Stock, and (5) junior to any series of our capital stock hereafter created specifically ranking by its terms senior to the Series F Preferred Stock in each case, as to dividends or distributions of assets upon our liquidation, dissolution or winding up whether voluntary or involuntary.
Conversion. Each share of the Series F Preferred Stock is convertible into shares of common stock at any time at the option of the holder, subject to the beneficial ownership limitation described below. The conversion rate of the Series F Preferred Stock is subject to proportionate adjustments for stock splits, reverse stock splits and similar events, but is not subject to adjustment based on price anti-dilution provisions.
Dividends. In addition to stock dividends or distributions for which proportionate adjustments will be made, holders of Series F Preferred Stock are entitled to receive dividends on shares of Series F Preferred Stock equal, on an as-if-converted-to-common-stock basis, to and in the same form as dividends actually paid on shares of the common stock when, as and if such dividends are paid on shares of the common stock. No other dividends are payable on shares of Series F Preferred Stock.
Voting Rights. Except as provided in the Certificate of Designation or as otherwise required by law, the holders of Series F Preferred Stock will have no voting rights. However, we may not, without the consent of holders of a majority of the outstanding shares of Series F Preferred Stock, alter or change adversely the powers, preferences or rights given to the Series F Preferred Stock, increase the number of authorized shares of Series F Preferred Stock, or enter into any agreement with respect to the foregoing.
Liquidation Rights. Upon any liquidation, dissolution or winding-up of the Company, whether voluntary or involuntary, the holders of Series F Preferred Stock are entitled to receive, pari passu with the holders of common stock, holders of Series A Convertible Preferred Stock, Series B Convertible Preferred Stock, Series C Convertible Preferred Stock, Series D Convertible Preferred Stock, and Series E Convertible Preferred Stock (on an as-converted basis), out of the assets available for distribution to stockholders, an amount equal to such amount per share as would have been payable had all shares of Series F Preferred Stock been converted into common stock immediately before such liquidation, dissolution or winding up, without giving effect to any limitation on conversion as a result of the Beneficial Ownership Limitation, as described below.
59
Beneficial Ownership Limitation. We may not effect any conversion of the Series F Preferred Stock, and a holder does not have the right to convert any portion of the Series F Preferred Stock to the extent that, after giving effect to the conversion set forth in a notice of conversion such holder would beneficially own in excess of the Beneficial Ownership Limitation, or such holder, together with such holder’s affiliates, and any persons acting as a group together with such holder or affiliates, would beneficially own in excess of the Beneficial Ownership Limitation. The “Beneficial Ownership Limitation” is 4.99% (or, at the election of the holder, of the number of shares of the common stock outstanding immediately after giving effect to the issuance of shares of common stock issuable upon conversion of Series F Preferred Stock held by the applicable holder. A holder may, with 61 days prior notice to us, elect to increase or decrease the Beneficial Ownership Limitation; provided, however, that in no event may either the holder Beneficial Ownership Limitation or the affiliate Beneficial Ownership Limitation be 9.99% or greater.
Exchange Listing. We do not plan on making an application to list the shares of Series F Preferred Stock on The Nasdaq Capital Market, any national securities exchange or other nationally recognized trading system. Our common stock issuable upon conversion of the Series F Preferred Stock is listed on The Nasdaq Capital Market.
Failure to Deliver Conversion Shares. If we fail to timely deliver shares of common stock upon conversion of the Series F Preferred Stock (the “Conversion Shares”) within the time period specified in the Certificate of Designation (within two trading days after delivery of the notice of conversion, or any shorter standard settlement period in effect with respect to trading market on the date notice is delivered), and if the holder has not exercised its Buy-In rights as described below with respect to such shares, then we are obligated to pay to the holder, as liquidated damages, an amount equal to $50 per business day (increasing to $100 per business day after the second business day and $200 per business day after the tenth business day) for each $5,000 of Conversion Shares for which the Series F Preferred Stock converted which are not timely delivered. If we make such liquidated damages payments, we are not also obligated to make Buy-In payments with respect to the same Conversion Shares.
Compensation for Buy-In on Failure to Timely Deliver Shares. If we fail to timely deliver the Conversion Shares to the holder, and if after the required delivery date the holder is required by its broker to purchase (in an open market transaction or otherwise) or the holder or its brokerage firm otherwise purchases, shares of common stock to deliver in satisfaction of a sale by the holder of the Conversion Shares which the holder anticipated receiving upon such conversion or exercise (a “Buy-In”), then we are obligated to (A) pay in cash to the holder the amount, if any, by which (x) the holder’s total purchase price (including brokerage commissions, if any) for the shares of common stock so purchased, minus any amounts paid to the holder by us as liquidated damages for late delivery of such shares, exceeds (y) the amount obtained by multiplying (1) the number of Conversion Shares that we were required to deliver times (2) the price at which the sell order giving rise to such purchase obligation was executed, and (B) at the option of the holder, either reinstate the portion of the Series F Preferred Stock and equivalent number of Conversion Shares for which such conversion was not honored (in which case such conversion shall be deemed rescinded) or deliver to the holder the number of shares of common stock that would have been issued had we timely complied with its conversion and delivery obligations.
Subsequent Rights Offerings; Pro Rata Distributions. If we grant, issue or sell any common stock equivalents pro rata to the record holders of any class of shares of common stock (the “Purchase Rights”), then a holder of Series F Preferred Stock will be entitled to acquire, upon the terms applicable to such Purchase Rights, the aggregate Purchase Rights which the holder could have acquired if the holder had held the number of shares of common stock acquirable upon conversion of the Series F Preferred Stock (without regard to any limitations on conversion). If we declare or make any dividend or other distribution of its assets (or rights to acquire its assets) to holders of common stock, then a holder of Series F Preferred Stock is entitled to participate in such distribution to the same extent as if the holder had held the number of shares of common stock acquirable upon complete conversion of the Series F Preferred Stock (without regard to any limitations on conversion).
Fundamental Transaction. If, at any time while the Series F Preferred Stock is outstanding, (i) we, directly or indirectly, in one or more related transactions effect any merger or consolidation of us with or into another person, (ii) we, directly or indirectly, effects any sale, lease, license, assignment, transfer, conveyance or other disposition of all or substantially all of our assets in one or a series of related transactions, (iii) any, direct or
60
indirect, purchase offer, tender offer or exchange offer (whether by us or another person) is completed pursuant to which holders of common stock are permitted to sell, tender or exchange their shares for other securities, cash or property and has been accepted by the holders of 50% or more of the outstanding common stock, (iv) we, directly or indirectly, in one or more related transactions effect any reclassification, reorganization or recapitalization of the common stock or any compulsory share exchange pursuant to which the common stock is effectively converted into or exchanged for other securities, cash or property, or (v) we, directly or indirectly, in one or more related transactions consummate a stock or share purchase agreement or other business combination (including, without limitation, a reorganization, recapitalization, spin-off or scheme of arrangement) with another person whereby such other person acquires more than 50% of the outstanding shares of common stock (not including any shares of common stock held by the other person or other persons making or party to, or associated or affiliated with the other persons making or party to, such stock or share purchase agreement or other business combination) (each a “Fundamental Transaction”), then the Series F Preferred Stock automatically converts and the holder will receive, for each share that would have been issuable upon such conversion immediately prior to the occurrence of such Fundamental Transaction (without regard to the Beneficial Ownership Limitation), the number of shares of common stock of the successor or acquiring corporation or of us, if we are the surviving corporation, and any additional consideration (the “Alternate Consideration”) receivable as a result of such Fundamental Transaction by a holder of the number of shares of common stock for which the Series F Preferred Stock is convertible immediately prior to such Fundamental Transaction (without regard to the Beneficial Ownership Limitation). For purposes of any such conversion, the determination of the conversion ratio will be appropriately adjusted to apply to such Alternate Consideration based on the amount of Alternate Consideration issuable in respect of one share of common stock in such Fundamental Transaction. If holders of common stock are given any choice as to the securities, cash or property to be received in a Fundamental Transaction, then the holder will be given the same choice as to the Alternate Consideration it receives upon automatic conversion of the Series F Preferred Stock following such Fundamental Transaction.
Warrants
As of May 30, 2023, there were warrants to purchase 1,613,483 shares of our common stock outstanding. The previously issued warrants all have a weighted average exercise price of $16.33 per warrant and have expiration dates between September 23, 2023 and February 3, 2028.
The following is a brief summary of the material terms of the warrants to purchase common stock offered pursuant to this prospectus and is subject in all respects to the provisions contained in the warrants, the form of which is filed as an exhibit to this prospectus. Pursuant to a warrant agency agreement between us and VStock Transfer, LLC, as warrant agent, the warrants will be issued in book-entry form and shall initially be represented only by one or more global warrants deposited with the warrant agent, as custodian, on behalf of The Depository Trust Company, or DTC, and registered in the name of Cede & Co., a nominee of DTC, or as otherwise directed by DTC.
Exercisability. The Class C Warrants are exercisable upon issuance and will expire on the date that is five years after their issuance date. The Class D Warrants are exercisable upon issuance and will expire on the date that is one year after their issuance date.The warrants are exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed exercise notice accompanied by payment in full for the number of shares of our common stock purchased upon such exercise (except in the case of a cashless exercise discussed below). The holder of warrants does not have the right to exercise any portion of the warrant if the holder would beneficially own in excess of 4.99% of the shares of our common stock outstanding immediately after giving effect to such exercise. This percentage may, however, be raised or lowered to an amount not to exceed 9.99% at the option of the holder upon at least 61 days’ prior notice from the holder to us.
Cashless Exercise. At any time when a registration statement covering the issuance of the shares of common stock issuable upon exercise of the warrants is not effective, the holder may, at its option, exercise its warrants on a cashless basis. When exercised on a cashless basis, a portion of the warrant is cancelled in payment of the purchase price payable in respect of the number of shares of our common stock purchasable upon such exercise.
61
Exercise Price. The exercise price of common stock purchasable upon exercise of the warrants is $ per share. The warrants will include a one-time reset of the exercise price to a price equal to the lesser of (i) the then exercise price and (ii) 90% of the five-day volume weighted average prices for the five (5) trading days immediately preceding the date that is sixty calendar days after issuance of the warrants. The exercise price and the number of shares issuable upon exercise of the warrants is subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications or similar events affecting our common stock. Holders of the warrants are entitled to participate in any subsequent rights offering or distribution of our assets on an as-if-exercised basis.
Transferability. The warrants may be transferred at the option of the holder upon surrender of the warrants with the appropriate instruments of transfer.
Exchange Listing. We do not plan on making an application to list the Class C Warrants or the Class D Warrants on The Nasdaq Capital Market, any national securities exchange or other nationally recognized trading system. Our common stock underlying the warrants is listed on The Nasdaq Capital Market.
Fundamental Transactions. The warrants provide that in the event of certain enumerated fundamental transactions, following such event, the holders of the warrants will be entitled to receive upon exercise of such warrants the same kind and amount of securities, cash or property which the holders would have received had they exercised their warrants immediately prior to such fundamental transaction. Any successor to us or surviving entity shall assume the obligations under the warrants. Additionally, as more fully described in the warrants, in the event of certain fundamental transactions, the holders of the warrants will be entitled to receive consideration in an amount equal to the Black Scholes value of the warrants on the date of consummation of such transaction.
Rights as Stockholder. Except as otherwise provided in the warrants (such as the rights described above of a warrant holder upon our sale or grant of any rights to purchase stock, warrants or securities or other property to our stockholders on a pro rata basis) or by virtue of such holder’s ownership of shares of our common stock, the holders of the warrants do not have the rights or privileges of holders of our common stock, including any voting rights, until they exercise their warrants.
Fractional Shares. No fractional shares of common stock will be issued upon the exercise of the warrants. Rather, the number of shares of common stock to be issued will be rounded up to the nearest whole number.
Anti-Takeover Effects of Delaware Law and Our Certificate of Incorporation and Bylaws
Some provisions of Delaware law, our restated certificate of incorporation and our amended and restated bylaws contain provisions that could make the following transactions more difficult: an acquisition of us by means of a tender offer; an acquisition of us by means of a proxy contest or otherwise; or the removal of our incumbent officers and directors. It is possible that these provisions could make it more difficult to accomplish or could deter transactions that stockholders may otherwise consider to be in their best interest or in our best interests, including transactions which provide for payment of a premium over the market price for our shares.
These provisions, summarized below, are intended to discourage coercive takeover practices and inadequate takeover bids. These provisions are also designed to encourage persons seeking to acquire control of us to first negotiate with our board of directors. We believe that the benefits of the increased protection of our potential ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure us outweigh the disadvantages of discouraging these proposals because negotiation of these proposals could result in an improvement of their terms.
Undesignated Preferred Stock. The ability of our board of directors, without action by the stockholders, to issue up to 10,000,000 shares of undesignated preferred stock with voting or other rights or preferences as designated by our board of directors could impede the success of any attempt to change control of us. These and other provisions may have the effect of deferring hostile takeovers or delaying changes in control or management of our company.
62
Stockholder Meetings. Our amended and restated bylaws provide that a special meeting of stockholders may be called only by our chairman of the board or chief executive officer (or president, if there is no chief executive officer), or by a resolution adopted by a majority of our board of directors.
Requirements for Advance Notification of Stockholder Nominations and Proposals. Our amended and restated bylaws establish advance notice procedures with respect to stockholder proposals to be brought before a stockholder meeting and the nomination of candidates for election as directors, other than nominations made by or at the direction of the board of directors or a committee of the board of directors.
Elimination of Stockholder Action by Written Consent. Our restated certificate of incorporation and amended and restated bylaws eliminate the right of stockholders to act by written consent without a meeting.
Staggered Board. Our board of directors is divided into three classes. The directors in each class serve for a three-year term, one class being elected each year by our stockholders. This system of electing and removing directors may tend to discourage a third-party from making a tender offer or otherwise attempting to obtain control of us, because it generally makes it more difficult for stockholders to replace a majority of the directors.
Removal of Directors. Our restated certificate of incorporation provides that no member of our board of directors may be removed from office by our stockholders except for cause and, in addition to any other vote required by law, upon the approval of not less than two-thirds (2/3) of the total voting power of all of our outstanding voting stock then entitled to vote in the election of directors.
Stockholders Not Entitled to Cumulative Voting. Our restated certificate of incorporation does not permit stockholders to cumulate their votes in the election of directors. Accordingly, the holders of a majority of the outstanding shares of our common stock entitled to vote in any election of directors can elect all of the directors standing for election, if they choose, other than any directors that holders of our preferred stock may be entitled to elect.
Delaware Anti-Takeover Statute. We are subject to Section 203 of the Delaware General Corporation Law, which prohibits persons deemed to be “interested stockholders” from engaging in a “business combination” with a publicly held Delaware corporation for three years following the date these persons become interested stockholders unless the business combination is, or the transaction in which the person became an interested stockholder was, approved in a prescribed manner or another prescribed exception applies. Generally, an “interested stockholder” is a person who, together with affiliates and associates, owns, or within three years prior to the determination of interested stockholder status did own, 15% or more of a corporation’s voting stock. Generally, a “business combination” includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. The existence of this provision may have an anti-takeover effect with respect to transactions not approved in advance by the board of directors.
Amendment of Charter Provisions. The amendment of any of the above provisions, except for the provision making it possible for our board of directors to issue preferred stock, would require approval by holders of at least 66 2/3% of the total voting power of all of our outstanding voting stock.
The provisions of Delaware law, our restated certificate of incorporation and our amended and restated bylaws could have the effect of discouraging others from attempting hostile takeovers and, as a consequence, they may also inhibit temporary fluctuations in the market price of our common stock that often result from actual or rumored hostile takeover attempts. These provisions may also have the effect of preventing changes in the composition of our board and management. It is possible that these provisions could make it more difficult to accomplish transactions that stockholders may otherwise deem to be in their best interests.
Transfer Agent and Registrar
The transfer and warrant agent and registrar for our common stock is VStock Transfer, LLC.
Listing
Shares of our common stock are quoted on The Nasdaq Capital Market under the symbol “KPRX.”
63
UNDERWRITING
We are offering the securities described in this prospectus through the underwriters named below. We have entered into an underwriting agreement dated , 2023 with Ladenburg Thalmann & Co. Inc. as the representative of the underwriters in this offering. Subject to the terms and conditions of the underwriting agreement, the underwriters have agreed to purchase the number of our securities set forth opposite its name below.
| Underwriters | Number of Shares of Common Stock (or Common Stock underlying Series F Preferred Stock) |
Number of Warrants |
||||||||||||
| Ladenburg Thalmann & Co. Inc. | ||||||||||||||
| Total | ||||||||||||||
A copy of the underwriting agreement has been filed as an exhibit to the registration statement of which this prospectus is part.
We have been advised by the underwriters that they propose to offer the shares of common stock and shares of Series F Preferred Stock, Class C Warrants and Class D Warrants directly to the public at the public offering price set forth on the cover page of this prospectus. Any securities sold by the underwriters to securities dealers will be sold at the public offering price less a selling concession not in excess of $ per share of common stock (or per share of common stock underlying Series F Preferred Stock) and $ per warrant.
The underwriting agreement provides that the underwriters’ obligation to purchase the securities we are offering is subject to conditions contained in the underwriting agreement.
No action has been taken by us or the underwriters that would permit a public offering of the securities in any jurisdiction outside the United States where action for that purpose is required. None of our securities included in this offering may be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sales of any of the securities offering hereby be distributed or published in any jurisdiction except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons who receive this prospectus are advised to inform themselves about and to observe any restrictions relating to this offering of securities and the distribution of this prospectus. This prospectus is neither an offer to sell nor a solicitation of any offer to buy the securities in any jurisdiction where that would not be permitted or legal.
The underwriters have advised us that they do not intend to confirm sales to any account over which they exercise discretionary authority.
Underwriting Discount and Expenses
The following table summarizes the underwriting discount and commission to be paid to the underwriters by us.
Per Share and Warrant (1)
|
Total Without Over-Allotment |
Total With Full Over-Allotment |
|||||||||||||||
| Public offering price | $ | ||||||||||||||||
Underwriting discounts and commissions to be paid to underwriters by us(2)(3)
|
$ | ||||||||||||||||
| Proceeds, before expenses, to us | $ | ||||||||||||||||
__________________
(1)The public offering price and underwriting discount corresponds, in respect of the securities (i) a public offering price per share of common stock (or per share of common stock underlying Series F Preferred Stock) of $ ($ net of the underwriting discount) and (ii) a public offering price per warrant of $ ($ net of the underwriting discount).
64
(2)We have also agreed to reimburse the accountable expenses of the representative, including a pre-closing expense allowance of up to a maximum of $25,000 and an additional closing expense allowance up to a maximum of $80,000.
(3)We have granted a 45-day option to the underwriters to purchase up to 387,324 additional shares of common stock, Class C Warrants exercisable for up to an additional 387,324 shares of common stock and/or Class D Warrants exercisable for up to an additional 387,324 shares of common stock at the assumed public offering price per share of common stock and the assumed public offering price per warrant set forth above less the underwriting discounts and commissions solely to cover over-allotments, if any.
We estimate the total expenses payable by us for this offering to be approximately , which amount includes (i) the underwriting discount of , (ii) reimbursement of the accountable expenses of the underwriters, including the legal fees of the representative, in an amount not to exceed $25,000 for pre-closing expenses plus $80,000 for closing expenses and (iii) other estimated company expenses of approximately which includes legal, accounting, and printing costs and various fees associated with the registration and listing of our shares.
The securities we are offering are being offered by the underwriters subject to certain conditions specified in the underwriting agreement.
Over-allotment Option
We have granted to the underwriters an option exercisable not later than 45 days after the date of this prospectus to purchase up to an additional 387,324 shares of common stock, Class C Warrants to purchase up to an additional 387,324 shares of common stock, and/or Class D Warrants to purchase up to an additional 387,324 shares of common stock at the assumed public offering price per share of common stock and the assumed public offering price per warrant set forth on the cover page hereto less the underwriting discounts and commissions. The underwriters may exercise the option solely to cover overallotments, if any, made in connection with this offering. If any additional shares of common stock and/or warrants are purchased, the underwriters will offer these shares and/or warrants on the same terms as those on which the other securities are being offered.
Right of First Refusal
We have granted to Ladenburg Thalmann & Co. Inc. the right of first refusal for a period of six months following the closing of this offering to act as sole bookrunner, exclusive placement agent or exclusive sales agent in connection with any financing of the Company, subject to certain conditions.
Listing
Our shares of common stock are listed on The Nasdaq Capital Market under the symbol “KPRX.”
The last reported sales price of our shares of common stock on May 30, 2023 was $2.13 per share. The actual public offering price will be determined between us, the underwriters and the investors in the offering, and may be at a discount to the current market price of our common stock. Therefore, the assumed public offering price used throughout this prospectus may not be indicative of the final public offering price. There is no established public trading market for the Series F Preferred Stock or the warrants, and we do not expect such markets to develop. In addition, we do not intend to apply for a listing of the Series F Preferred Stock, the Class C Warrants or the Class D Warrants on any national securities exchange or other nationally recognized trading system.
Lock-up Agreements
Our officers, directors, each of their respective affiliates and associated partners have agreed with the underwriters to be subject to a lock-up period of 90 days following the date of this prospectus. This means that, during the applicable lock-up period, such persons may not offer for sale, contract to sell, sell, distribute, grant any option, right or warrant to purchase, pledge, hypothecate or otherwise dispose of, directly or indirectly, any shares of our common stock or any securities convertible into, or exercisable or exchangeable for, shares of our common stock. Certain limited transfers are permitted during the lock-up period if the transferee agrees to these lock-up restrictions. We have also agreed, in the underwriting agreement, to similar lock-up restrictions on the issuance and sale of our securities for 90 days following the closing of this offering, although we will be permitted to issue stock options or stock awards to directors, officers and employees under our existing plans. Ladenburg Thalmann & Co. Inc. may, in their sole discretion and without notice, waive the terms of any of these lock-up agreements.
65
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is VStock Transfer, LLC.
Determination of Offering Price
Our common stock is currently traded on The Nasdaq Capital Market under the symbol “KPRX.” On May 30, 2023 the closing price of our common stock was $2.13 per share. We do not intend to apply for listing of the Series F Preferred Stock, the Class C Warrants or the Class D Warrants on any securities exchange or other trading system.
The public offering price of the securities offered by this prospectus will be determined by negotiation between us and the underwriters. Among the factors that will be considered in determining the public offering price:
•our history and our prospects;
•the industry in which we operate;
•our past and present operating results;
•the previous experience of our executive officers; and
•the general condition of the securities markets at the time of this offering.
The assumed combined public offering price stated on the cover page of this prospectus should not be considered an indication of the actual value of the shares of common stock, shares of preferred stock or warrants sold in this offering. That price is subject to change as a result of market conditions and other factors and we cannot assure you that the shares of common stock sold in this offering can be resold at or above the public offering price.
Stabilization, Short Positions and Penalty Bids
The underwriters may engage in syndicate covering transactions stabilizing transactions and penalty bids or purchases for the purpose of pegging, fixing or maintaining the price of our common stock:
•Syndicate covering transactions involve purchases of securities in the open market after the distribution has been completed in order to cover syndicate short positions. Such a naked short position would be closed out by buying securities in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the securities in the open market after pricing that could adversely affect investors who purchase in the offering.
•Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specific maximum.
•Penalty bids permit the underwriters to reclaim a selling concession from a syndicate member when the securities originally sold by the syndicate member are purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions.
These syndicate covering transactions, stabilizing transactions, and penalty bids may have the effect of raising or maintaining the market prices of our securities or preventing or retarding a decline in the market prices of our securities. As a result the price of our common stock may be higher than the price that might otherwise exist in the open market. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of our common stock. These transactions may be effected on The Nasdaq Capital Market, in the over-the-counter market or on any other trading market and, if commenced, may be discontinued at any time.
In connection with this offering, the underwriters also may engage in passive market making transactions in our common stock in accordance with Regulation M during a period before the commencement of offers or sales of shares of our common stock in this offering and extending through the completion of the distribution. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for that security.
66
However, if all independent bids are lowered below the passive market maker’s bid that bid must then be lowered when specific purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
Neither we, nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the prices of our securities. In addition, neither we nor the underwriters make any representation that the underwriters will engage in these transactions or that any transactions, once commenced will not be discontinued without notice.
Other Relationships
From time to time, certain of the underwriters and their affiliates may provide in the future, various advisory, investment and commercial banking and other services to us in the ordinary course of business, for which they will receive customary fees and commissions. The representative has acted as book-running manager in connection with our public offering that we consummated in July 2022 and as warrant solicitation agent in connection with a warrant inducement transaction that we consummated in November 2022 for which it received compensation.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including certain liabilities arising under the Securities Act, or to contribute to payments that the underwriters may be required to make for these liabilities.
67
LEGAL MATTERS
Certain legal matters with respect to the validity of the securities offered by this prospectus will be passed upon for us by Burns & Levinson LLP, Boston, MA. Ellenoff Grossman & Schole LLP, New York, New York, is acting as counsel to the underwriters in connection with this offering.
EXPERTS
The consolidated balance sheets of Kiora Pharmaceuticals, Inc. as of December 31, 2022 and 2021, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the years then ended, have been audited by EisnerAmper LLP, independent registered public accounting firm, as stated in their report dated March 23, 2023, which is incorporated herein by reference, which report includes an explanatory paragraph about the existence of substantial doubt concerning the Company’s ability to continue as a going concern. Such financial statements have been incorporated herein by reference in reliance on the report of such firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the securities being offered by this prospectus. This prospectus does not contain all of the information in the registration statement and its exhibits. For further information with respect to us and the common stock offered by this prospectus, we refer you to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract or any other document referred to are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference.
We are subject to the information requirements of the Exchange Act and, in accordance therewith, file annual, quarterly and special reports, proxy statements and other information with the SEC. These documents may be accessed through the SEC’s electronic data gathering, analysis and retrieval system, or EDGAR, via electronic means, including the SEC’s home page on the Internet (www.sec.gov).
We post on our public website (www.kiorapharma.com) our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Our website and the information contained on that site, or connected to that site, are not incorporated into and are not a part of this prospectus.
68
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The SEC allows us to incorporate by reference the information and reports we file with it under File No. 001-36672, which means that we can disclose important information to you by referring you to those publicly available documents. The information incorporated by reference is an important part of this prospectus, and information that we file later with the SEC will automatically update and supersede the information already incorporated by reference. We are incorporating by reference the documents listed below:
•Our Annual Report on Form 10-K for the year ended December 31, 2022 filed with the SEC on March 23, 2023;
•Our Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2023 filed with the SEC on May 9, 2023;
•Our Current Reports on Form 8-K filed with the SEC on January 3, 2023, February 3, 2023, February 7, 2023, March 30, 2023 and April 27, 2023 (in each case, except for information contained therein which is furnished rather than filed);
•The description of our common stock contained in our registration statement on Form 8-A12B filed with the SEC on July 28, 2015 and amended on July 30, 2015; and
•All future documents filed by us with the SEC pursuant to Section 13(a), 13(c), 14 or 15(d) of the Exchange Act, including all filings made after the date of the filing of this registration statement and prior to the effectiveness of this registration statement, prior to the termination of the offering of the underlying securities; provided, however, that we are not incorporating by reference any additional documents or information furnished and not filed with the SEC.
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus is modified or superseded for purposes of the prospectus to the extent that a statement contained in this prospectus or in any other subsequently filed document that also is or is deemed to be incorporated by reference herein modifies or supersedes such statement.
Upon request, we will provide, without charge, to each person, including any beneficial owner, to whom a copy of this prospectus is delivered a copy of the documents incorporated by reference into this prospectus. You may request a copy of these filings, and any exhibits we have specifically incorporated by reference as an exhibit in this prospectus, at no cost by writing or telephoning us at the following address:
| Kiora Pharmaceuticals, Inc. | ||
| 332 Encinitas Boulevard, Suite 102 | ||
| Encinitas, California 92024 | ||
| Telephone: (858) 224-9600 | ||
69
2,582,160 Shares of Common Stock
5,500 Shares of Series F Convertible Preferred Stock
(2,582,160 shares of Common Stock underlying the Series F
Convertible Preferred Stock)
Class C Warrants to Purchase up to 2,582,160 Shares of Common Stock
(2,582,160 shares of Common Stock underlying the Warrants)
and Class D Warrants to Purchase up to 2,582,160 Shares of Common Stock
(2,582,160 shares of Common Stock underlying the Warrants)
PROSPECTUS
Ladenburg Thalmann
, 2023
70
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth all expenses to be paid by us in connection with this offering, other than underwriting discounts and commissions. All amounts shown are estimates except for the SEC registration fee and FINRA filing fee.
Securities and Exchange Commission registration fee |
$ | 2,851.44 | |||
FINRA filing fee |
$ | 3,200.00 | |||
Legal fees and expenses |
$ | 75,000.00 | |||
Accounting fees and expenses |
$ | 98,250.00 | |||
Transfer agent fees and expenses |
$ | 3,000.00 | |||
Printing expenses |
$ | 8,000.00 | |||
Miscellaneous |
$ | 15,000.00 | |||
| Total | $ | 205,301.44 | |||
Item 14. Indemnification of Directors and Officers.
Our amended and restated certificate of incorporation contains provisions that eliminate, to the maximum extent permitted by the General Corporation Law of the State of Delaware, the personal liability of our directors for monetary damages for breach of their fiduciary duties as directors. Our amended and restated bylaws provide that we must indemnify our directors and officers and may indemnify our employees and other agents to the fullest extent permitted by the General Corporation Law of the State of Delaware.
Sections 145 and 102(b)(7) of the General Corporation Law of the State of Delaware provide that a corporation may indemnify any person made a party to an action by reason of the fact that he or she was a director, officer, employee or agent of the corporation or is or was serving at the request of a corporation against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by him or her in connection with such action if he or she acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful, except that, in the case of an action by or in right of the corporation, no indemnification may generally be made in respect of any claim as to which such person is adjudged to be liable to the corporation.
We have entered into indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our amended and restated bylaws, and intend to enter into indemnification agreements with any new directors and executive officers in the future. We have purchased and intend to maintain insurance on behalf of any person who is or was a director or officer of us against any loss arising from any claim asserted against him or her and incurred by him or her in any such capacity, subject to certain exclusions. See also “Undertakings” set out in response to Item 17 herein.
Item 15. Recent Sales of Unregistered Securities.
Set forth below is information regarding the shares of common stock and preferred stock and the warrants issued, and options granted, by us in the three years preceding the filing of this registration statement that were not registered under the Securities Act.
(1)On December 18, 2020, we entered into a Share Purchase Agreement with the shareholders of Panoptes Pharma Ges.m.b.H (“Panoptes”). Pursuant to the agreement, we acquired all of the outstanding equity interests of Panoptes, and Panoptes became a wholly-owned subsidiary of the Company. The consideration paid by us to the former shareholders of Panoptes at closing in connection with the acquisition, after adjustment as provided in the Share Purchase Agreement and including consideration paid to the sellers’
II-1
financial advisor, was comprised of (i) 22,106 shares of the common stock, (ii) 45.8923 shares of Series D Convertible Preferred convertible into an aggregate of 325 shares of common stock, and (iii) cash payments in an aggregate amount of approximately $220,577. Additionally, 1,500 shares of Series D Preferred Stock convertible into an aggregate of approximately 10,617 shares of common stock may be issued after a period of 18 months subject to adjustments for potential post-closing working capital and/or indemnification claims relating to breaches of representations, warranties and covenants contained in the Share Purchase Agreement. The Series D Convertible Preferred Stock has a stated value of $1,000 per share and a conversion price of $141.284 per share. The Series D Convertible Preferred Stock is only entitled to dividends in the event dividends are paid on shares of common stock and will not have any preferences over shares of common stock or any voting rights, except in limited circumstances.
(2)On January 5, 2021, we entered into a Securities Purchase Agreement with Armistice Capital Master Fund, Ltd. (the “Investor”), pursuant to which we agreed to issue to the Investor in a private placement 38,278 shares of common stock and warrants to purchase 38,278 shares of common stock for an aggregate purchase price of approximately $8.0 million. The private placement closed on January 6, 2021. The warrants have an exercise price of $209 per share and became exercisable on the six-month anniversary of their issuance date. The warrants are exercisable for five years from the issuance date.
(3)On August 9, 2021, we entered into a Securities Purchase Agreement with several institutional and accredited investors for the sale of 116,721 shares common stock in a registered direct offering. Concurrently with the sale of the shares, we also sold to the investors unregistered warrants to purchase up to an aggregate of 58,361 shares of common stock. The gross proceeds to us from the offerings were approximately $10.75 million, before deducting the placement agent’s fees and other offering expenses, and excluding the proceeds, if any, from the exercise of the warrants. Subject to certain ownership limitations, the warrants were immediately exercisable upon issuance at an exercise price equal to $89.60 per share of common stock, subject to adjustments as provided under the terms of the warrants. The warrants are exercisable for five and one-half years from the initial exercise date. The closing occurred on August 11, 2021. We agreed to pay H.C. Wainwright & Co., LLC, the placement agent for the offerings, an aggregate cash fee equal to 7% of the aggregate gross proceeds received from the offerings plus a management fee equal to 1% of the aggregate gross proceeds received from the sale of the securities in the offerings. In addition, we also agreed to issue to the placement agent or its designees warrants to purchase up to 5,836 shares of common stock. The warrants have an exercise price of $115.124 per share of common stock and will be exercisable for five years from the commencement of the sales pursuant to the offering.
(4)On October 21, 2021, we entered into a Stock Purchase Agreement with the former stockholders of Bayon Therapeutics, Inc. (“Bayon”). Pursuant to the agreement, we acquired all of the outstanding equity interests of Bayon, and Bayon became our wholly-owned subsidiary of the Company. The consideration paid by us to the former Bayon stockholders at closing in connection with the acquisition, after adjustment as provided in the agreement, was comprised of 845 shares of common stock. In addition to the consideration set forth above, the former Bayon stockholders are eligible to receive up to $7.1 million in additional payments based on clinical trial and FDA approval milestones for Bayon’s product candidates, as set forth in the agreement. In each case, we may elect to pay the applicable milestone payment either (i) in cash, or (ii) by issuing shares of common stock, provided that we may not issue shares of common stock under the agreement that, in the aggregate, exceed 5% of the number of shares of common stock outstanding immediately prior to the consummation of the acquisition unless approval of the Company’s stockholders is obtained.
(5)On November 17, 2022, we entered into Inducement Letters with certain investors from our August 2022 public offering pursuant to which such investors agreed to exercise for cash all of their Class A Warrants to purchase 654,609 shares of our common stock originally issued in the public offering in exchange for our agreement to issue new inducement warrants on substantially the same terms as the Class A Warrants, except as described below, to purchase up to 654,609 shares of common stock, in the Private Placement. Each inducement warrant is exercisable at a price per share of common stock of $5.97. Each inducement warrant will initially be exercisable six months following its date of issuance, and will expire on the 18 month anniversary of their initial exercise date.
II-2
(6)On February 2, 2023, we entered into a securities purchase agreement with Lincoln Park Capital, LLC (“Lincoln Park”) whereby we issued 52,798 shares of common stock and warrants to purchase 105,596 shares of common stock to Lincoln Park in a private placement. The combined purchase price in the private placement was $3.788 per share and two warrants. Each warrant is exercisable at a price per share of common stock of $3.538, will initially be exercisable six months following its date of issuance, and will expire on the five year anniversary of its initial exercise date.
(7)On February 3, 2023, we entered into the Purchase Agreement with Lincoln Park, pursuant to which we have the right to sell to Lincoln Park up to $10.0 million in shares of common stock, subject to certain limitations, from time to time over the 36 month period commencing on the date that a registration statement covering the resale of the shares is declared effective by the SEC. As of May 30, 2023, we had sold 125,000 shares of common stock to Lincoln Park pursuant to the Purchase Agreement for aggregate gross proceeds of approximately $442,310.
The offers, sales and issuances of the securities described in this Item 15 were deemed to be exempt from registration under the Securities Act in reliance on Section 4(a)(2) of the Securities Act and Rule 506 promulgated under Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were affixed to the securities issued in these transactions.
Item 16. Exhibits and Financial Statement Schedules.
(a)Exhibits.
See the Exhibit Index immediately before the Signature Pages.
(b)Financial Statement Schedules.
All schedules have been omitted because they are either inapplicable or the required information has been given in the financial statements or notes thereto.
Item 17. Undertakings.
(a)Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
(b)The undersigned registrant hereby undertakes:
(1)To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i)To include any prospectus required by Section 10(a)(3) of the Securities Act;
(ii)To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered
II-3
(if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement;
(iii)To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(2)That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3)To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4)That, for the purpose of determining liability under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i)Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424 (§230.424 of this chapter);
(ii)Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii)The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv)Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
(5)That, for purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(6)That, for the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-4
EXHIBIT INDEX
| Exhibit Number | Description of Exhibit | |||||||
| 1.1* | ||||||||
| 2.1** | ||||||||
| 2.2** | ||||||||
| 2.3** | ||||||||
| 3.1 | ||||||||
| 3.2 | ||||||||
| 3.3 | ||||||||
| 3.4 | ||||||||
| 3.5 | ||||||||
| 3.6 | ||||||||
| 3.7 | ||||||||
| 3.8 | ||||||||
| 3.9 | ||||||||
| 3.10 | ||||||||
| 3.11 | ||||||||
| 3.12 | ||||||||
| 3.13*** | ||||||||
II-5
| 4.1 | ||||||||
| 4.2 | ||||||||
| 4.3 | ||||||||
| 4.4 | ||||||||
| 4.5 | ||||||||
| 4.6 | ||||||||
| 4.7 | ||||||||
| 4.8 | ||||||||
| 4.9 | ||||||||
| 4.10 | ||||||||
| 4.11 | ||||||||
| 4.12* | ||||||||
| 4.13* | ||||||||
| 4.14* | ||||||||
| 5.1* | ||||||||
| 10.1# | ||||||||
| 10.2# | ||||||||
| 10.3# | ||||||||
| 10.4 | ||||||||
| 10.5# | ||||||||
| 10.6# | ||||||||
II-6
| 10.7† | ||||||||
| 10.8 | ||||||||
| 10.9†† | ||||||||
| 10.10†† | ||||||||
| 10.11†† | ||||||||
| 10.12 | ||||||||
| 10.13 | ||||||||
| 10.14# | ||||||||
| 10.15†† | ||||||||
| 10.16†† | ||||||||
| 10.17# | ||||||||
| 10.18# | ||||||||
| 10.19# | ||||||||
| 10.20# | ||||||||
| 10.21 | ||||||||
| 10.22** | ||||||||
| 10.23 | ||||||||
II-7
| 10.24 | ||||||||
| 10.25** | ||||||||
| 16.1 | ||||||||
| 21.1 | ||||||||
| 23.1* | ||||||||
| 23.2* | ||||||||
| 24.1*** | ||||||||
| 107* | ||||||||
__________________
*Filed herewith.
** Schedules and exhibits have been omitted pursuant to Item 601(a)(5) of Regulation S-K. The registrant hereby undertakes to furnish copies of any of the omitted schedules and exhibits upon request by the U.S. Securities and Exchange Commission.
*** Previously filed.
† Confidential treatment requested as to portions of the exhibit. Confidential materials omitted and filed separately with the Securities and Exchange Commission.
†† Certain confidential portions of this exhibit were omitted because the identified confidential portions (i) are not material and (ii) would be competitively harmful if publicly disclosed.
# Management contract or compensatory plan or arrangement.
II-8
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this Amendment No. 2 to Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Encinitas, State of California, on this 31st day of May, 2023.
| KIORA PHARMACEUTICALS, INC. | |||||
| By: | /s/ Brian M. Strem, Ph.D. | ||||
| Brian M. Strem, Ph.D. | |||||
| President and Chief Executive Officer | |||||
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||||||||||||
| /s/ Brian M. Strem, Ph.D. |
President, Chief Executive Officer and Director (principal executive officer)
|
May 31, 2023 | ||||||||||||
| Brian M. Strem, Ph.D. | ||||||||||||||
| /s/ Melissa Tosca |
Executive Vice President of Finance
(principal financial and accounting officer)
|
May 31, 2023 | ||||||||||||
| Melissa Tosca | ||||||||||||||
| * | Chairman | May 31, 2023 | ||||||||||||
| Paul Chaney | ||||||||||||||
| * | Director | May 31, 2023 | ||||||||||||
| Kenneth Gayron | ||||||||||||||
| * | Director | May 31, 2023 | ||||||||||||
| Praveen Tyle | ||||||||||||||
| * | Director | May 31, 2023 | ||||||||||||
| David Hollander | ||||||||||||||
| * | Director | May 31, 2023 | ||||||||||||
| Aron Shapiro | ||||||||||||||
| * | Director | May 31, 2023 | ||||||||||||
| Erin Parsons | ||||||||||||||
*By:
/s/ Brian M. Strem, Ph.D.
Brian M. Strem, Ph.D.
Attorney-in-Fact
II-9